This is the second part of this thread: Duplicated genes and genome fragmentation
As @Michael G. suggested, I did a pseudo-annotation of sequences 5-2 and 5-3 (i.e., I joined both sequences into one) and I did the phylogenetic analysis again. Here is the tree that I obtained:
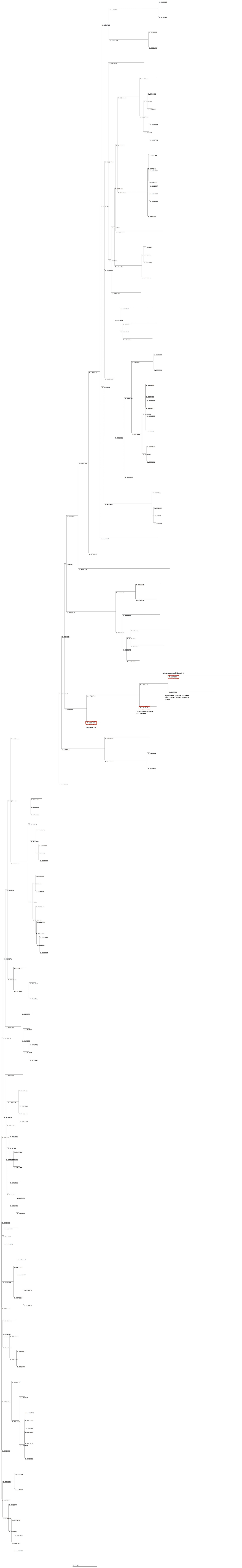
Both of my sequences are shown in a red rectangle. The length of the branch seems to have decreased a bit. Also, I looked at the branches near sequence 5-1. One of them is the original query sequence from species A and the other one is a hypothetical protein from the same species (probably related to the query sequence, but I couldn't find any domains because it is only a few amino acids long). I still need to find the root of the phylogeny but I am not sure how to do this...