I ran HiSat2, MarkDuplicate, removed reads with the lower quality score than 40 and finally only kept properly paired reads.
After the BAM filtering steps, I used the Scallop results with TransDecoder. Next, I used bamCoverage to create RNA-Seq profile. I noticed that not always the first isoform is the best one compared to the RNA-Seq profile as could be seen below a few examples:
1.

2.
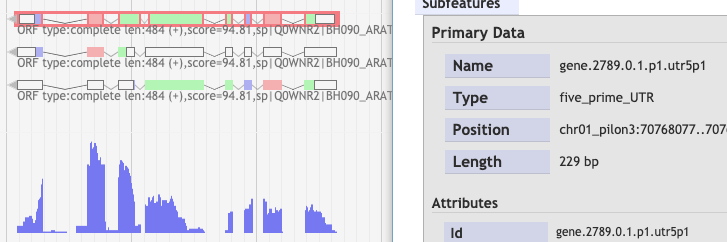
3.

4.

5.

By any chance, is there a way to keep only the best isoform which is in red box?
Thank you in advance,