Summary My genome tree doesn't agree with my gene trees and I get the feeling that my genome tree might be wrong, possibly due to long branch attraction, but I don't know how to check/fix it.
Background I have a set of genes of interest recovered from genomes and metagenomes. I also have some bins built from the metagenomes of interest.
Previous work I've built multiple gene trees, which tend to agree with each other, as well as with other phylogenetic methods (16S rRNA, initial genome trees where available, conserved orientation of the genes of interest). So at this point I though I had a pretty good idea of how my genes / species of interest are linked to the species tree.
Concern I've assembled the metagenomes and recovered bins. I've refined bins of interest with anvio, based on sequence composition / differential coverage, and kept only the bins which appeared quite high quality (>70% completion*, <10 - often <5% - redundancy). Thus contamination in the metagenome assemblies (MAGs) is unlikely. The contamination would have to happen multiple times in the same way to different samples from different environments, at the same coverage, to get the same effect.
- with the exception of a bin of particular interest which I accepted with 62%
I have de-replicated these bins, added some known genomes, and tried to build a species tree with Orthofinder (with the proteomes). There is a problem occurs with a clade of 3 bins I will call 'X'.
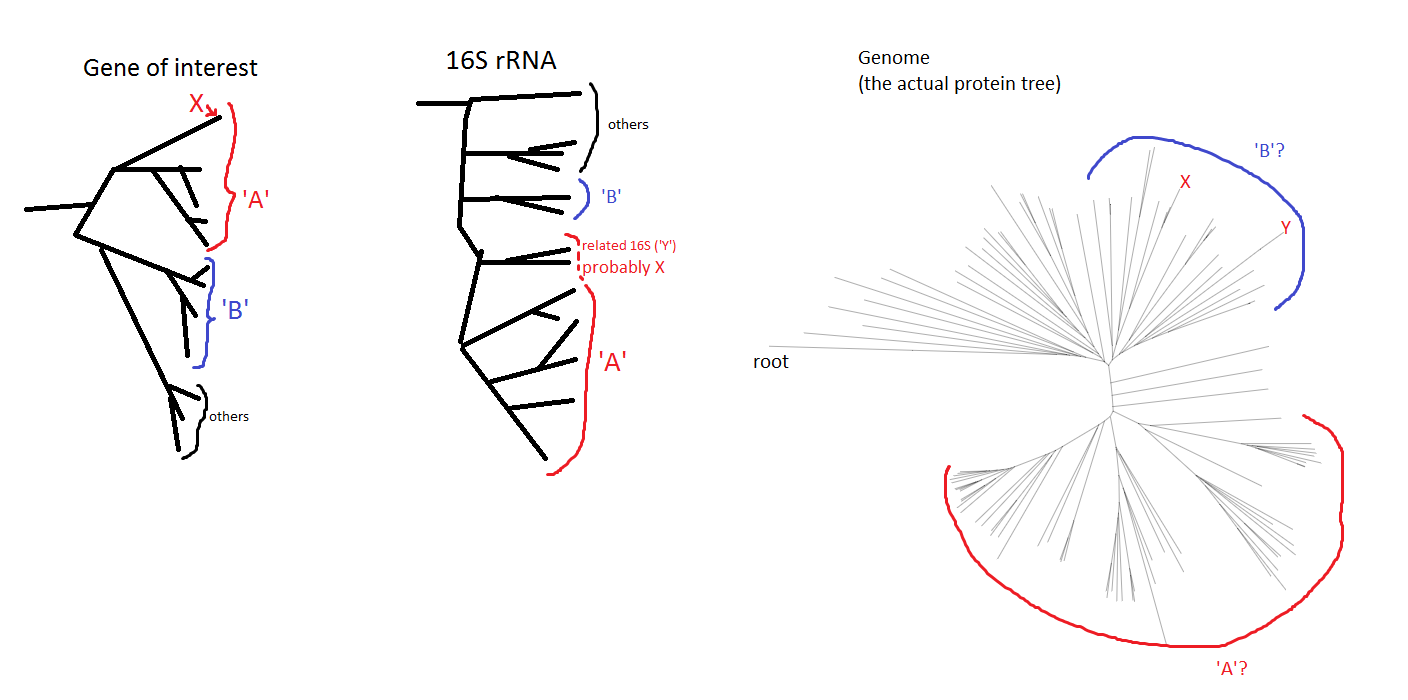
Trees Based on the gene-of-interest phylogenies, X looks like probably a sister-group to the rest of known genomes from order 'A'. Which is very interesting. With the 16S rRNA, it's not quite as clear (there are no 16S rRNA sequences in the bins); however, some 16S rRNA amplified from one of the original samples fit the bill as 'sister-clade to basically everything else in group A'). The first (100 or so) BLAST hits for this 16S sequence are all from group 'A', still.
Tree conflict However, in the proteome tree, X as well as Y (Y being a sequenced genome that I suspected was related to X based on 16S data) end up in order 'B' instead of 'A'. GTDBtk also labels X as belonging to clade 'B', not 'A'.
Now,I know that genes and genomes don't always have the same evolutionary trajectories. But multiple sets of data lead me to a different conclusion than the proteome tree, so I have to at least question whether the proteome tree is correct; what's likely to be the problem (I suspect long-branch attraction), and, if it is long-branch attraction (or anything else) how can I uncover the correct signal?
Thank you for your time!