I have a problem here with my rna seq data:
Sequencing details:
rRNA was removed, followed by cDNA preparation and generation of stranded libraries using the TruSeq Stranded Total RNA Sample Prep Kit. Sequencing performed on the HiSeq2500 platform (Illumina) to generate 2 × 125 bp paired-end reads
Alignment and preprocessing
reads were aligned using tophat2(std aligner in pipeline at core (--library-type fr-firststrand), unfortunately the original unaligned files were purged and i only have access to this aligned file which i converted to fastq using following steps
samtools sort -n sample.bam -o sample_sorted.bambedtools bamtofastq -i sample_sorted.bam -fq sample_1.fq -fq2 sample_2.fq(get a lot of mate skipping errors here)- check for these reads for adapters and trim them and again use hisat2 (with
--rna-strandness RF)
trimmed reads
Reads were trimmed by passing parameters --adapters adapters.file --adapter-trim-end RIGHT --length-dist --threads 12 --adapter-min-overlap 7 --max-uncalled 250 --min-read-length 25 -- to FLEXBAR version 2.4
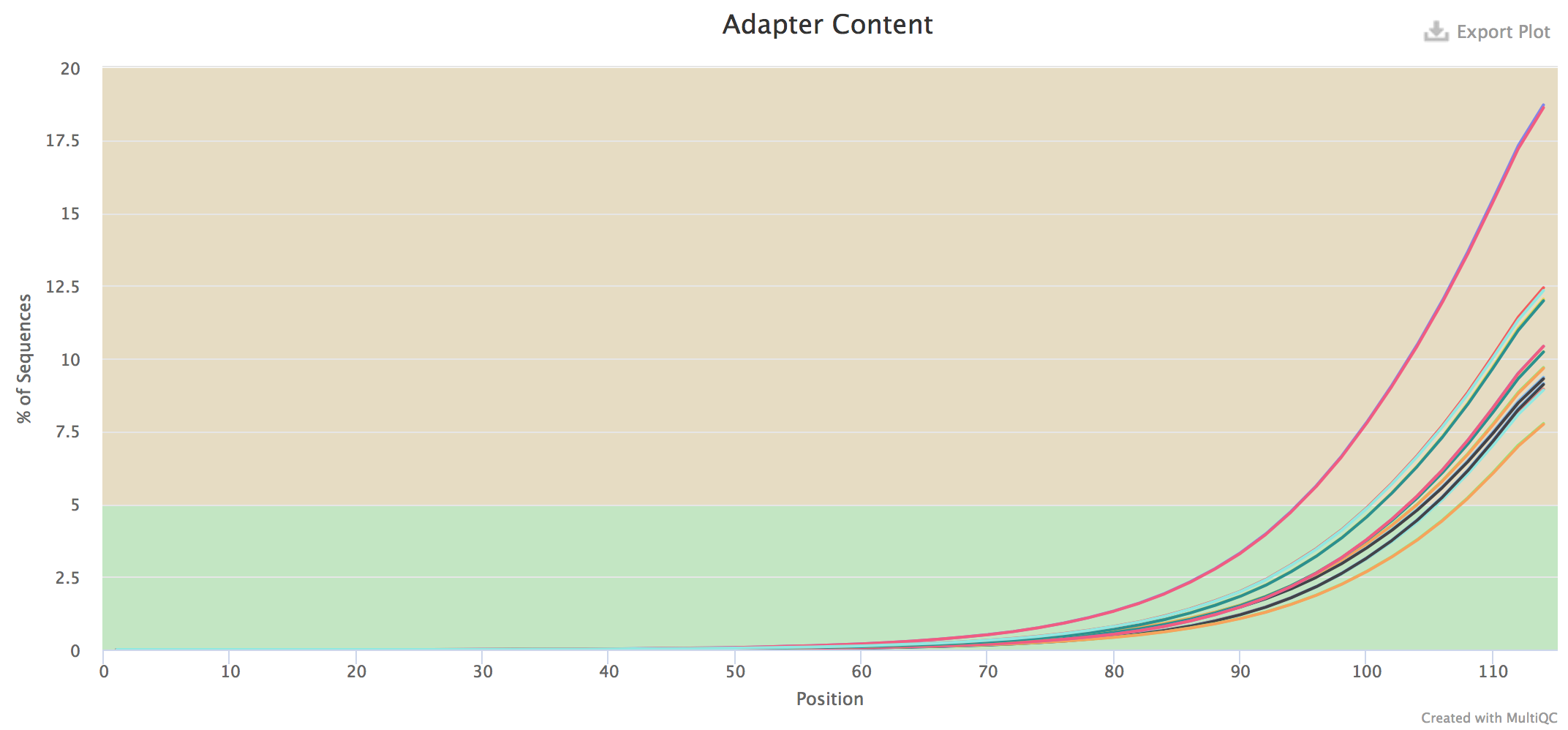
adapters for all samples using multiqc
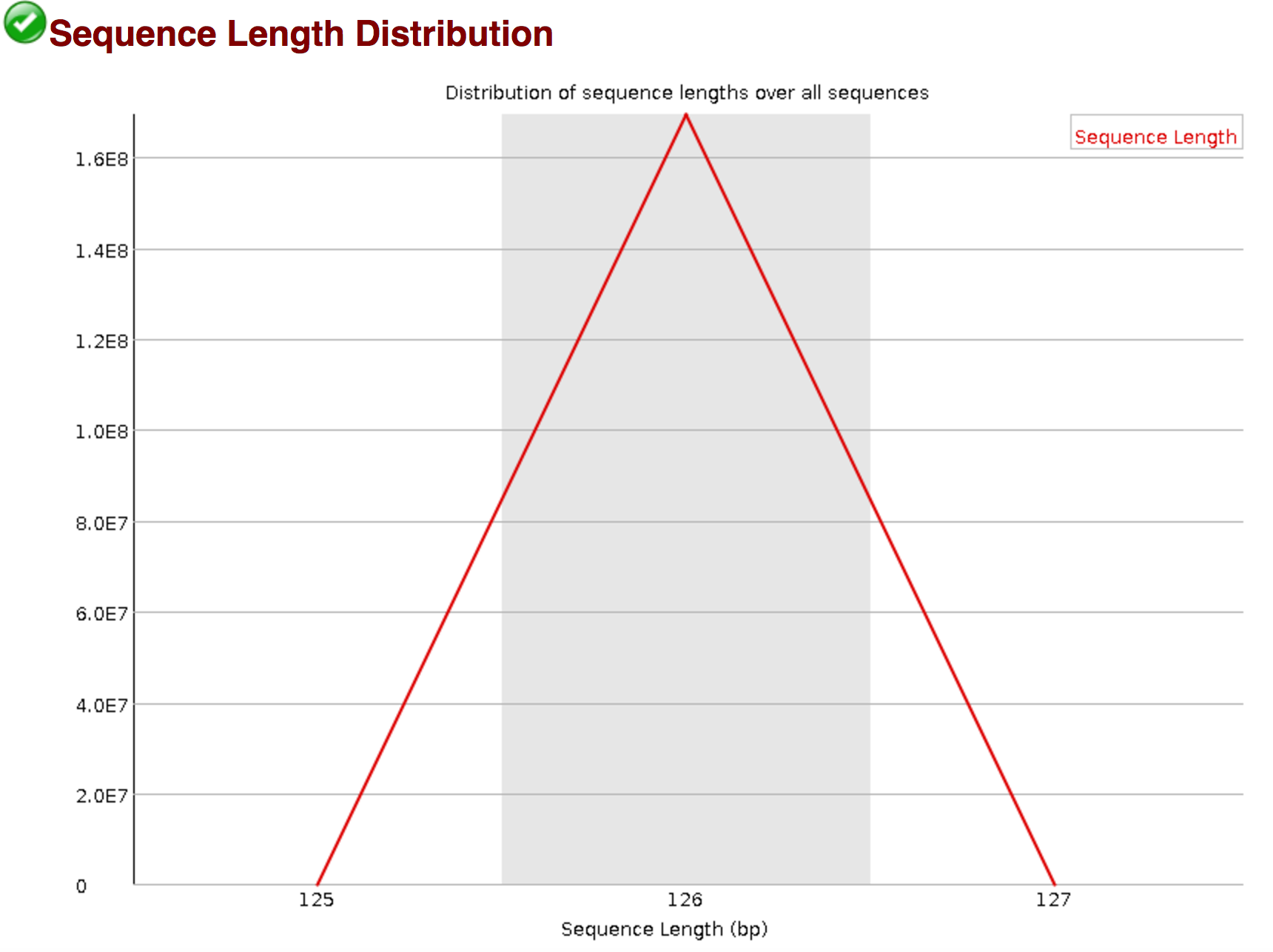
fastqc sequence length pre trimming
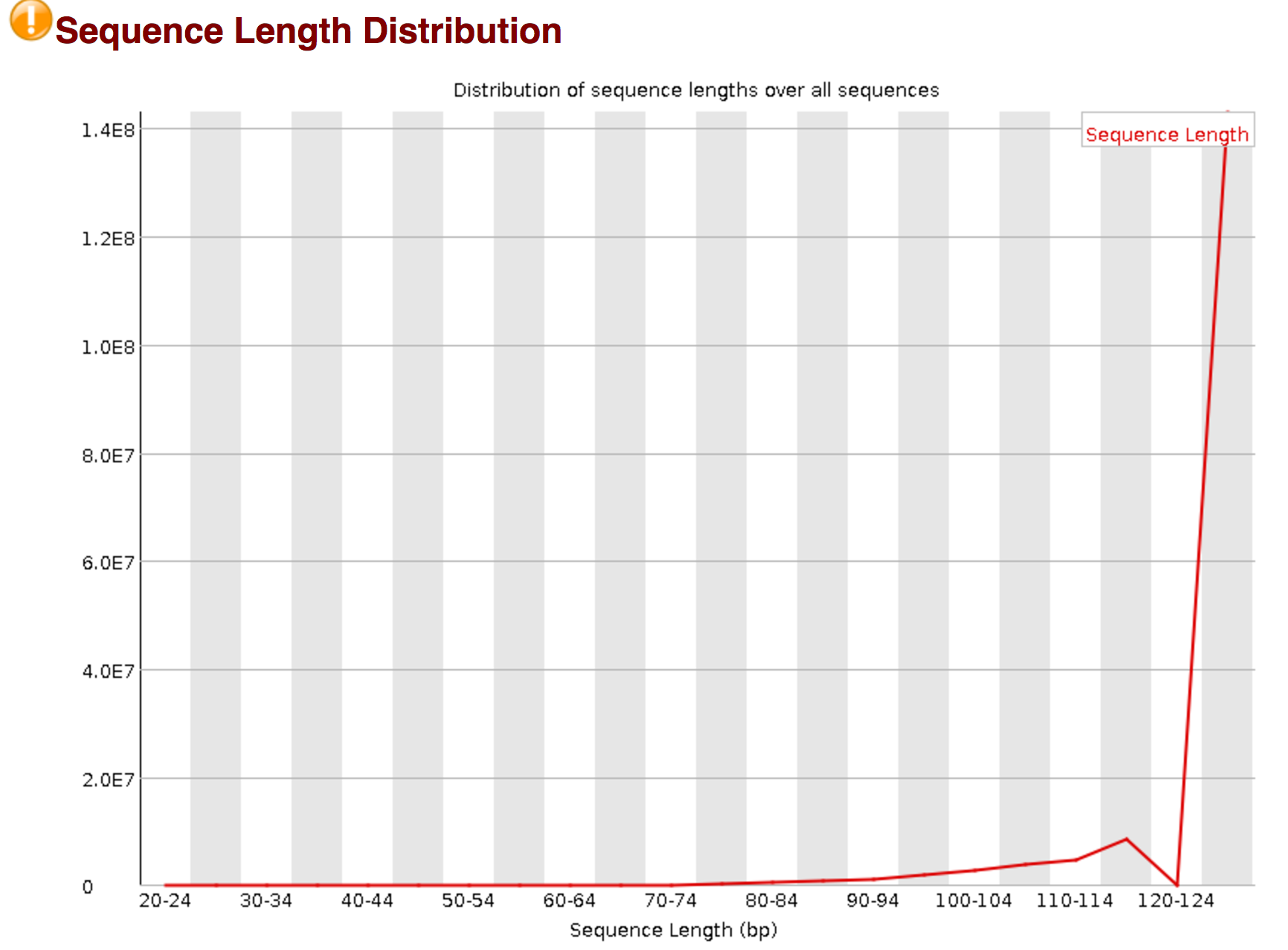
fastqc sequence length post trimming
rmats error
Incorrect readLength. sample.bam has a read length of 114, while readLength param is 125
picard error on aligned bam from hisat2)
ProcessExecutor 2 "Not creating insert size PDF as there are duplicated header names: All_Reads"
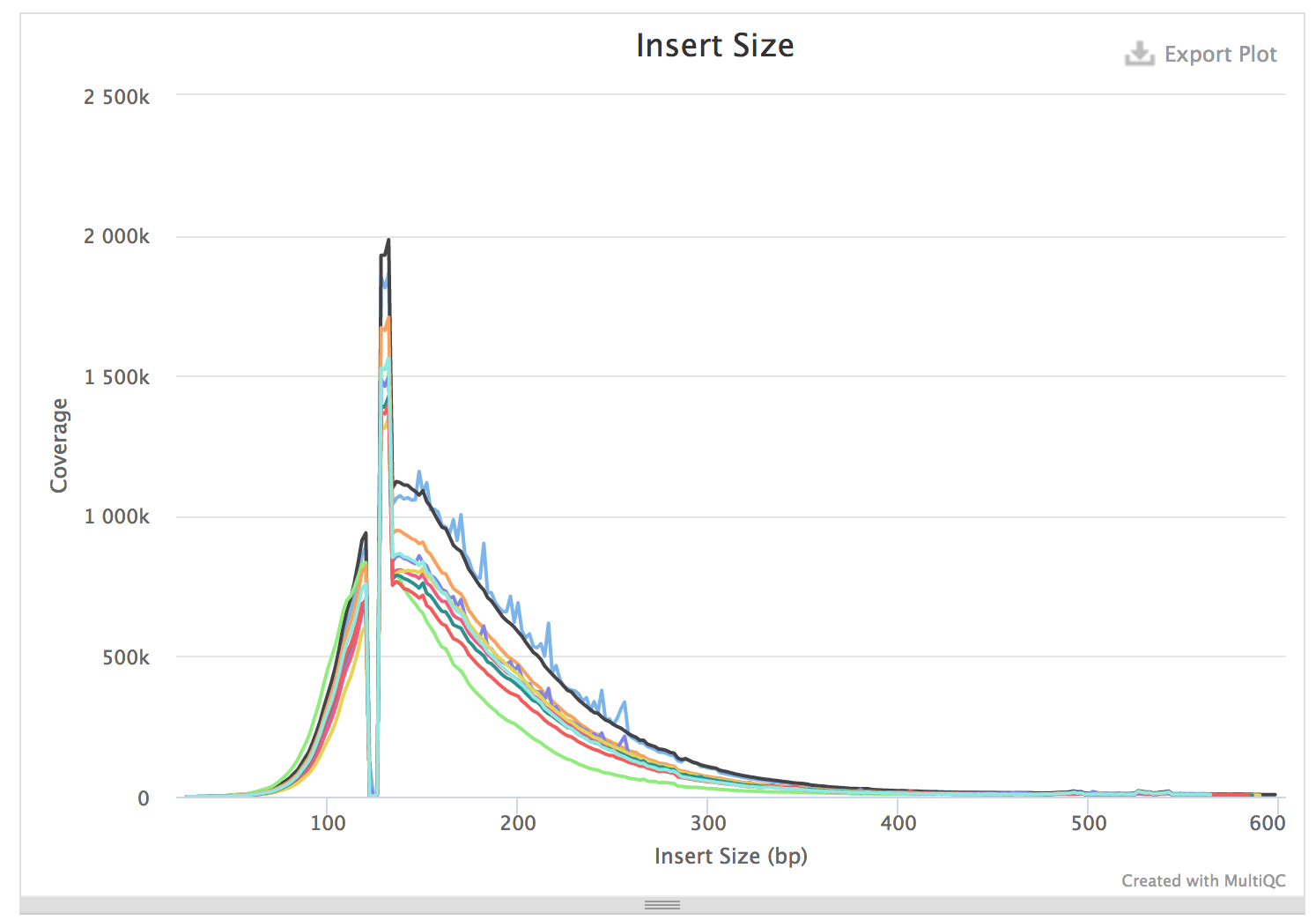
snapshot of picard insertsize metrics file
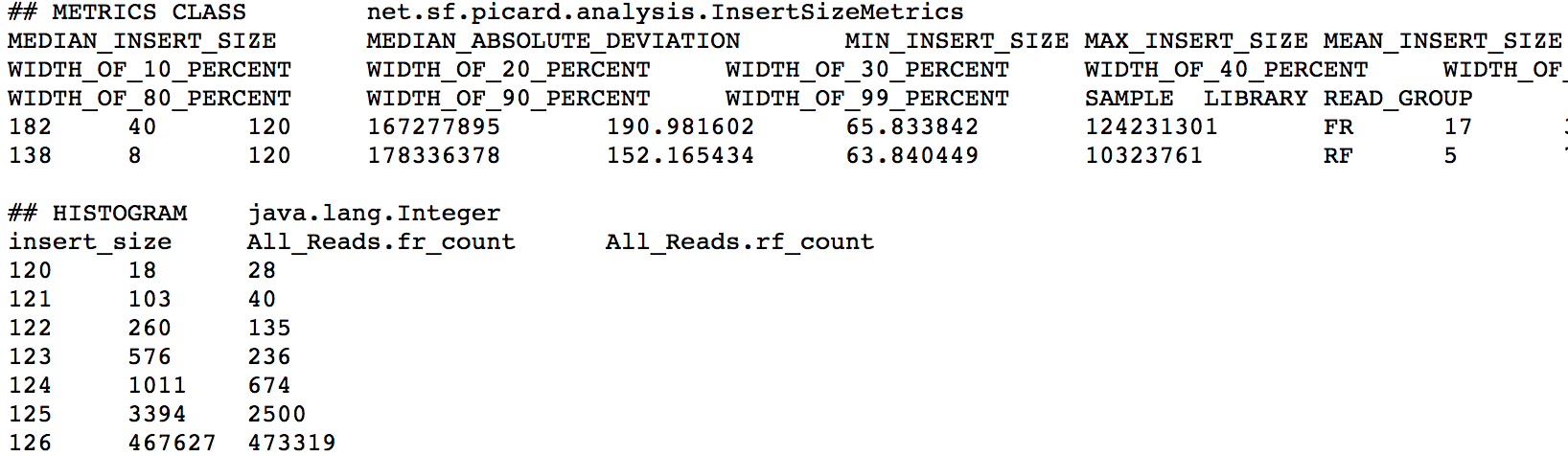
Questions
- Is my approach flawed at some step?
- why do i get different read lengths error for each sample when i process it through rmats when i specify len as 125?
- why is no histogram being produced when using picard insertsize metrics?
- is normal to see the graph shift to the right after trimming?
insertsize for all samples (data from picard collectinsertsize compiled using multiqc)