This question was also asked on Biostars
I am performing a de novo genome assembly using Illumina paired-end short reads, sequenced on a NovaSeq X by our collaborator at UCLA.
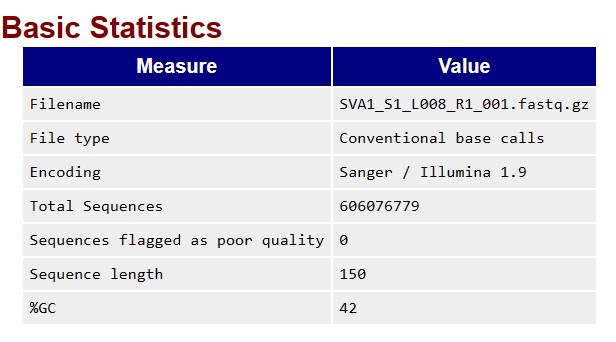
At present, I am in the stage of trimming the adapters. Here, you can have a look at the basic statistics and information on the adapter content obtained from the Fast QC report, for R1.
FOR RAW READS
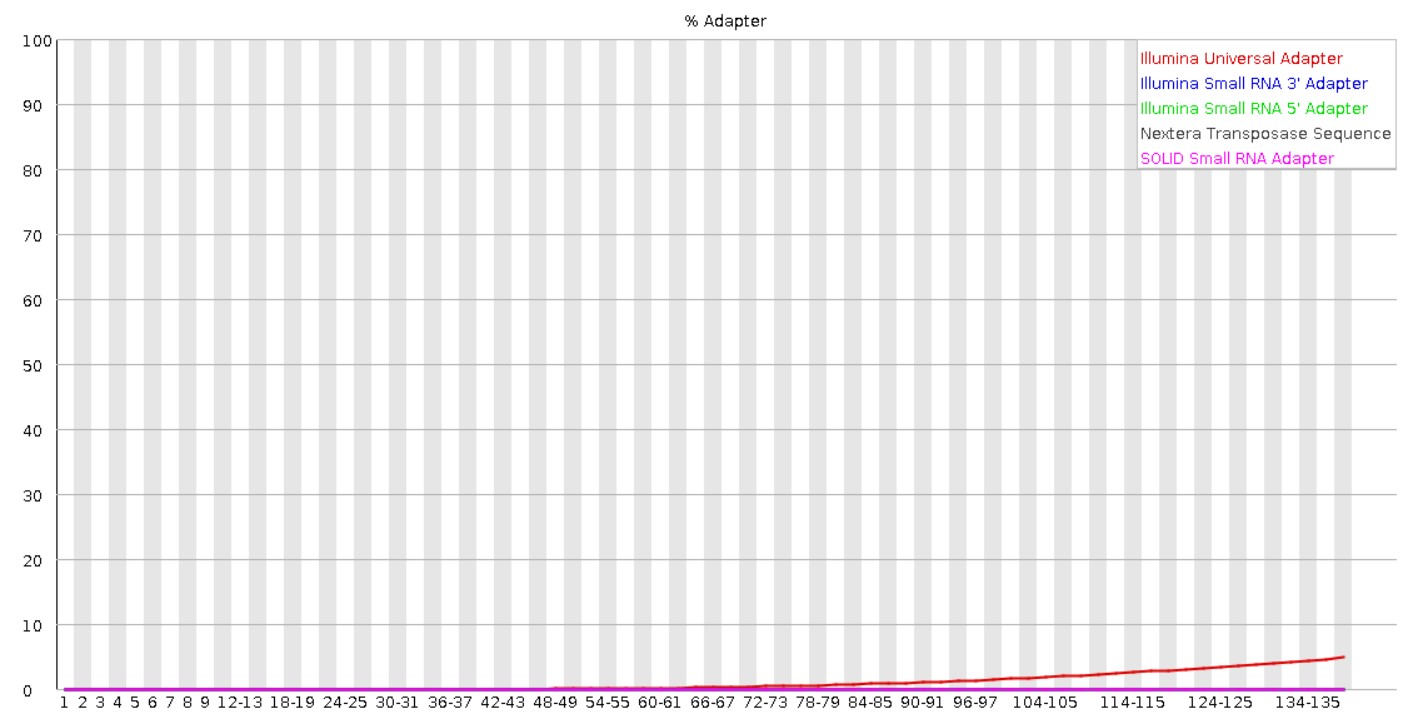
I used Trimmomatic for trimming the adapter. The following is the Trimmomatic Settings
ILLUMINACLIP:~/adapters/TruSeq3-PE.fa:2:30:10 MINLEN:36
Here, you can see the basic statistics and adapter content of the Trimmed reads.

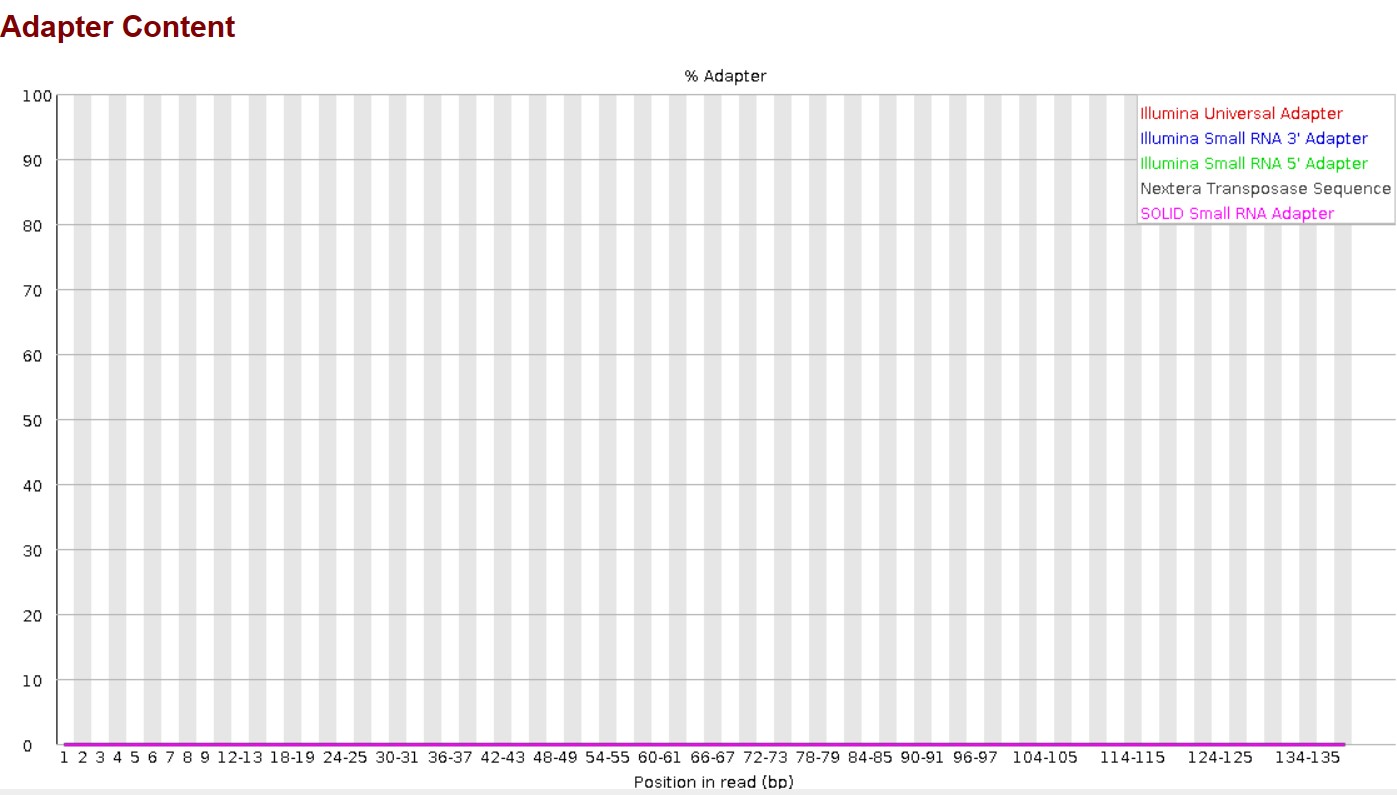
Here, the output was:
Both surviving: 566832403 Forward only surviving: 39244376 Reverse only surving: 0.00
Dropped reads: <1%
Here you can go through the full FastQC report for:
Now I have two questions:
Question 1
Can I go ahead with the assembly process, for there is zero-adapter-presence in the reads? Should I mind the loss of reads?
Question 2
I see that there are over-represented sequences, both in read 1 and read 2. I doubt if I can leave them be, or I should trim them too. I would like to know if the over-represented sequences need to be tossed, or if they can be left untouched and proceed with the assembly. If I need to remove these sequences, can Trimmomatic do it?
If I were to trim the over-represented sequences, can I use an adapter "fasta" file as follows?
The following are the over-represented sequences for R1.
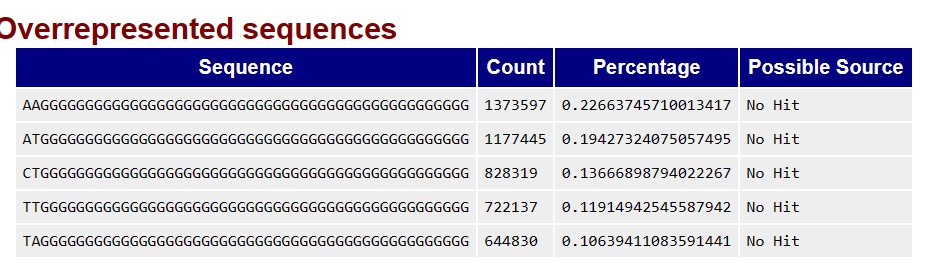
The following are the over-represented sequences for R2.
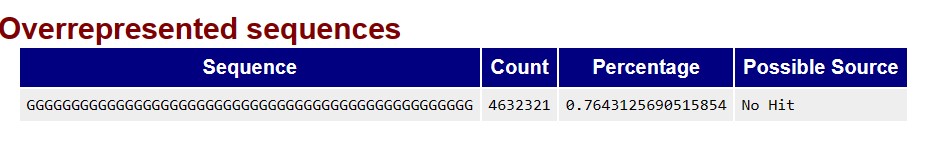
Can I have my fasta file as follows, to remove the over-represented sequences:
>PrefixPE/1
-------sequence1--------------
-------sequence2--------------
-------sequence3--------------
-------sequence4--------------
>PrefixPE/2
-------sequence1--------------
java -jar trimmomatic-0.30.jar PE s_1_1_sequence.txt.gz s_1_2_sequence.txt.gz
lane1_forward_paired.fq.gz lane1_forward_unpaired.fq.gz lane1_reverse_paired.fq.gz
lane1_reverse_unpaired.fq.gz ILLUMINACLIP:overRepresentedSeqs.fa:2:30:10 MINLEN:36
NB: Sorry about the lengthy post. Since the file sizes are large, it takes over a day for the Trimmomatic to finish the task, so that it is not feasible to go with multiple rounds of trial and error check.