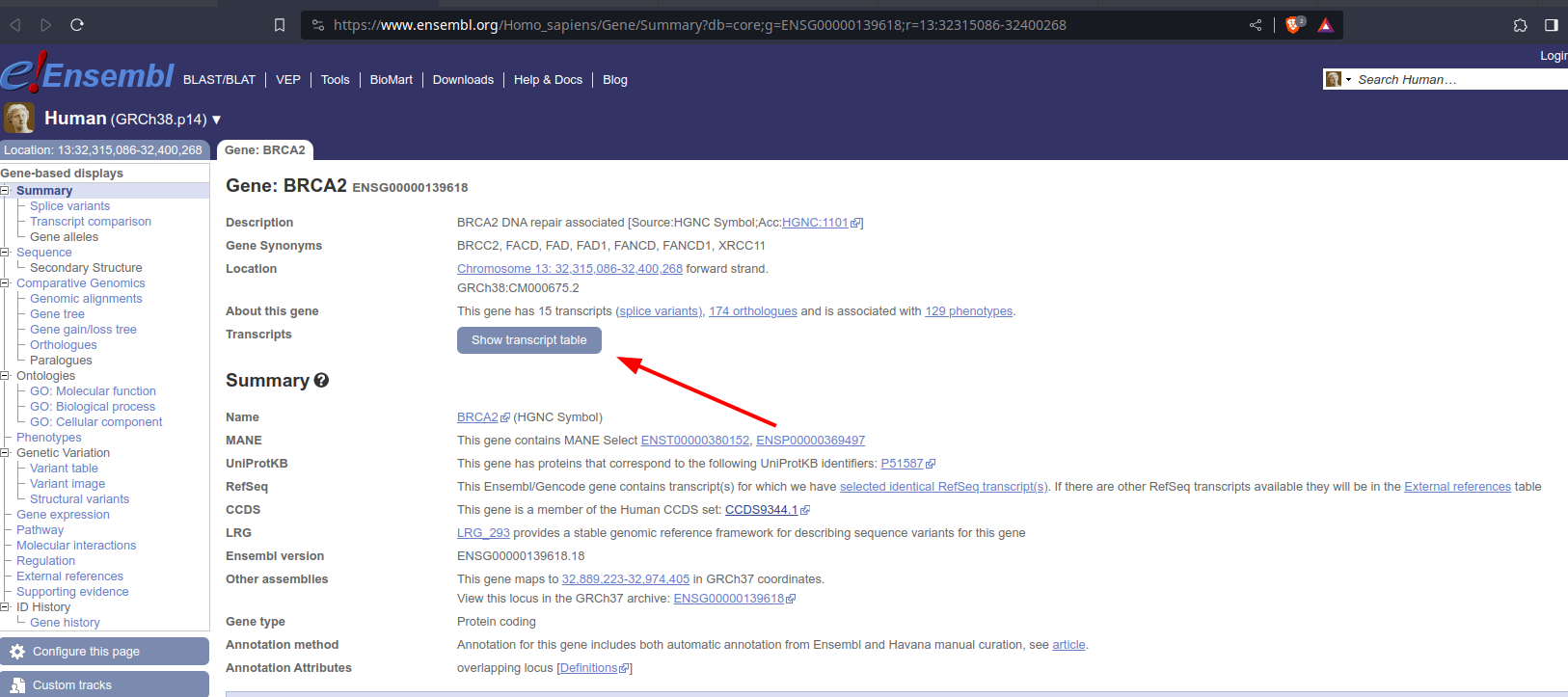
[Update] Thanks @terdon. To clarify my question:
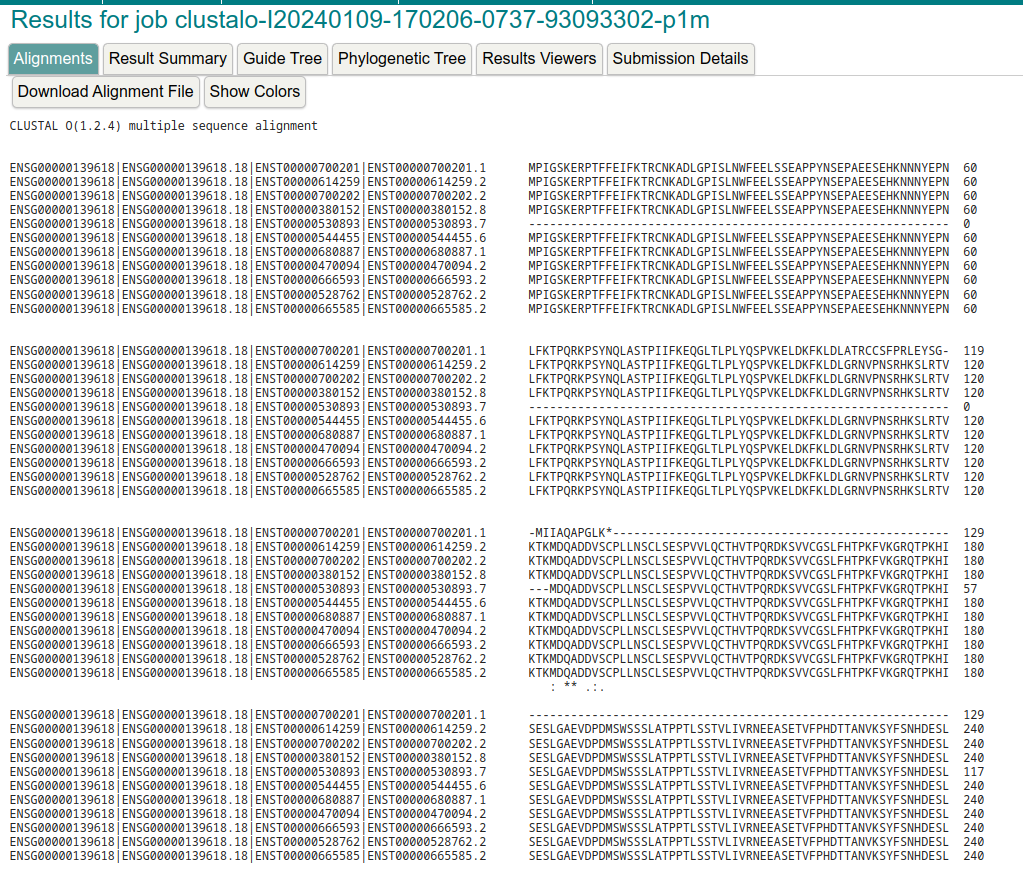
I have a bunch of protein isoforms sequences (produced by the transcripts in the figure) and I want to align protein products (e.g., proteins produced by the first 3 protein-coding transcripts against each other). See there are 3 blocks missing in 5' region in BRCA2-204 compared against BRCA2-206.
This case might be very easy with any MSA softwares such as clustal omega, but there are complicated cases which may trick software into aligning single amino acids instead of being splicing aware. Instead of manually adjusting the alignments, I want to know whether I can directly download, or "translate", this figure into alignment results (e.g., fasta format or clustal format)
This figure should be visible with https://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000139618;r=13:32315086-32400268 in the gene summary tab
[Original] Is there a way to directly download the sequence alignment result from Ensembl? If not, what will be the best way to align exons and introns without parameter-tuning