I am running a single cell dataset (count data - exon) through Seurat. After running tsne I see a cluster (13) split in 3 different locations on the plot. Here are the commands I am running:
library('Seurat')
## rows (genes) that have 99% zeros were filtered out,
## and then columns (cells) that have 99% zeros were filtered out
isp.data <- read.table(file = "genes.cells99.percent.filtered", sep =
",", row.names = 1, header=TRUE)
isp <- CreateSeuratObject(raw.data = isp.data, project =
"brain_regions")
isp <- NormalizeData(object = isp, normalization.method =
"LogNormalize",scale.factor = 10000)
isp <- FindVariableGenes(object = isp, mean.function = ExpMean,
dispersion.function = LogVMR,do.plot = FALSE)
hv.genes <- head(rownames([email protected]), 1000)
isp <- ScaleData(object = isp, genes.use = hv.genes, num.cores = 3)
isp <- RunPCA(object = isp, pc.genes = hv.genes, pcs.compute = 100,
do.print = TRUE, pcs.print = 1:5, genes.print = 5)
PCElbowPlot(object = isp, num.pc = 100)
isp <- JackStraw(object = isp, num.replicate = 100, display.progress =
FALSE, num.pc = 30)
JackStrawPlot(object = isp, PCs = 1:30)
isp <- FindClusters(object = isp, reduction.type = "pca", dims.use =
1:26, resolution= 0.75, print.output = 0, save.SNN = TRUE,
force.recalc=TRUE)
isp <- RunTSNE(isp, dims.use = 1:26, do.fast = T)
TSNEPlot(isp, do.label = T, pt.size = 0.5)
Here is the tsne plot:
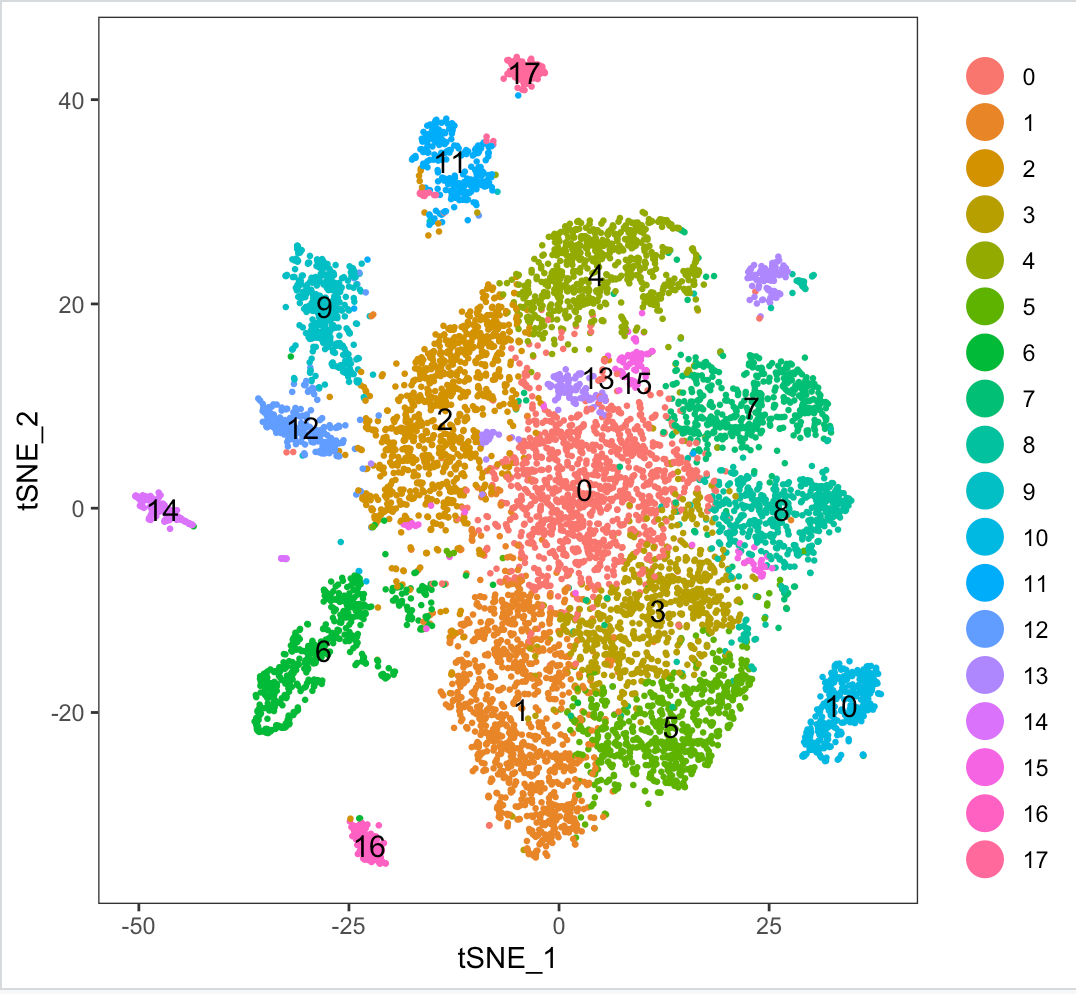
I have tried varying parameters for resolution in FindClusters (from 0.5-3.5), but that just increases the clusters found. Further I tried to play around with parameters min.dist (0.75,1), n_neighbors (10 to 30) and perplexity(5 to 50) but I still get at least 1-2 clusters split in the tsne plot.
Another thing I have tried is to reduce the dimension (dims.use=10) on which tsne is ran but the clusters seem to be too close together, like so:
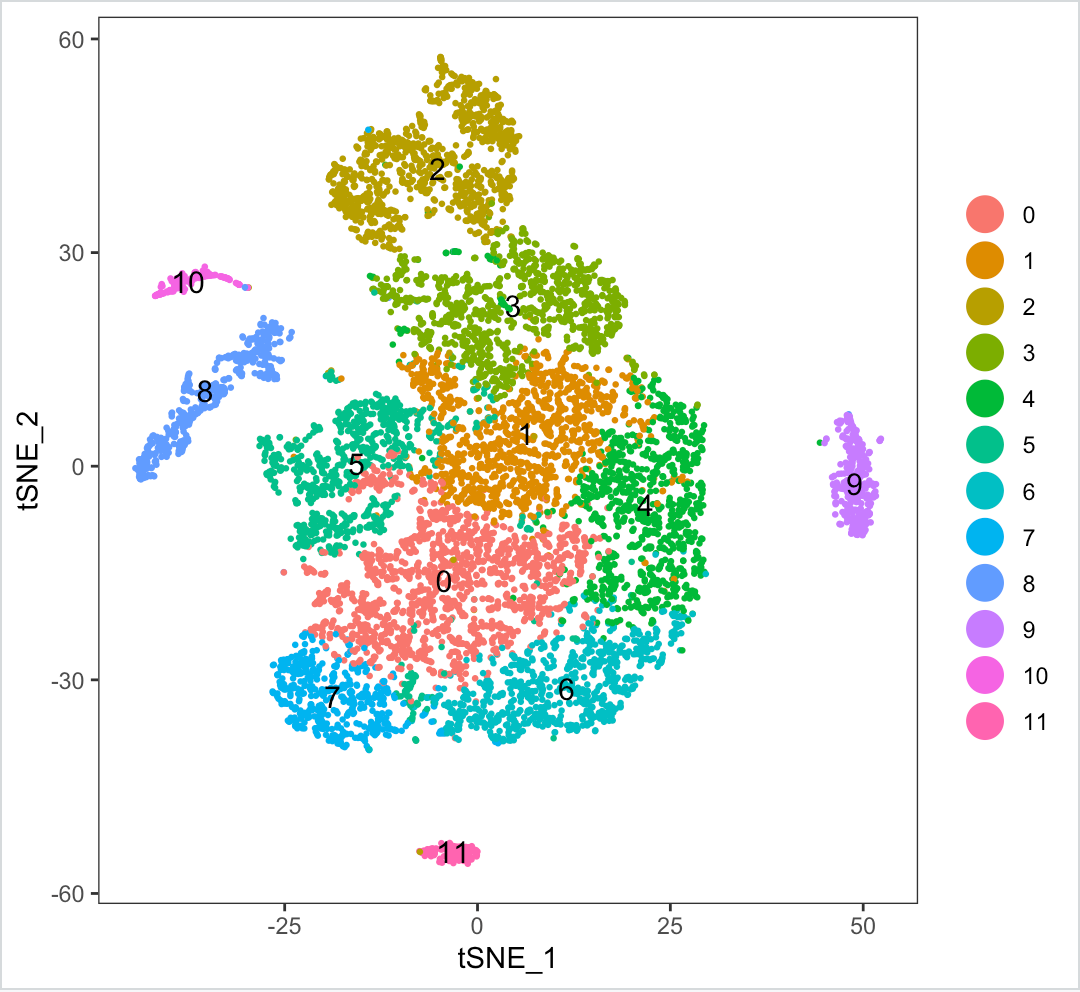
Note: Dimensionality of dataset (after filtering) - 23824 genes across 8967 samples.
My question being why does such behavior happen? Is my filtering strategy not correct? Have I not normalized/scaled data correctly?