I need to merge multiple channels each emits multiple file. So far I did something as below:
process prephasing {
publishDir params.out, mode:'copy'
input:
tuple val(prefix), path(bfiles) from plink_data
each chrom from chroms
path hrc_sites_file
output:
tuple val("${chrom}"),path("chr${chrom}*.{bed,bim,fam}") into qc1_chr
"""
PRE_PASHING_QC2.sh "${hrc_sites_file}" "${prefix}" "${chrom}"
"""
}
channel.fromPath('/some/path/genetic_map_chr*_combined_b37.txt').into {shape_map;i
mp_map}
channel.fromPath('/some/path/*hrc.r1-1.ega.grch*.hap.gz').into {shape_ref;
imp_ref}
channel.fromPath('/some/path/*hrc.r1-1.ega.grch37.chr*.legend.gz').into {s
hape_legend;imp_legend}
channel.fromPath('/some/path/*hrc.r1-1.ega.grch37.chr*.samples').into {sha
pe_sample;imp_sample}
qc1_chr.filter{name, bfiles -> bfiles.collect {it.getBaseName()}.every {it.endsWith("step10")}}.into {shape_in;imp_in}
shape_para=shape_in.merge(shape_map).merge(shape_ref).merge(shape_legend).merge(shape_sample)
process phasing {
publishDir params.out, mode:'copy'
input:
each file(penta) from shape_para
output:
path("*step10*") into phased
script:
def (in, map, ref, legend, sample) = penta
"""
shapeit2 -T 32 --input-bed "${in}" --input-map "${map}" --input-ref "${ref}" "${legend}" "${sample}" --duohmm -O "${in}.phased" --seed 54321
"""
}
The "prephasing" process works fine and produce expected output. But the "phasing" process does not produce any output nor any error. Any help?
I have tried according to @Steve as below:
channel.fromFilePairs('/cluster/projects/p697/projects/moba_qc_imputation/resources/1000Genomes/genetic_map_chr*_combined_b37.txt',size:1).map {group_key, file_list -> tuple(group_key.replaceFirst(/^genetic_map_/,""),file_list.first())}.set {genetic_map}
channel.fromFilePairs('/cluster/projects/p697/projects/moba_qc_imputation/resources/HRC/haplegendsample/*hrc.r1-1.ega.grch37.chr*.haplotypes.{hap.gz,legend.gz,samples}',size:3).map{group_key,file_list -> tuple(group_key.replaceFirst(/^*HRC\.r1-1\.ega\.grch37\./,""),file_list)}.set {ref_panel}
qc1_chr.filter{chrom, bfiles -> bfiles.collect {it.getBaseName()}.every {it.endsWith("step10")}}.join(ref_panel).join(genetic_map).into {shape_in;imp_in}
process phasing {
publishDir params.out, mode:'copy'
input:
tuple val(chrom), path(input_bed), path(intput_ref), path(input_map) from shape_in
output:
tuple val("${chrom}"),path("${chrom}.{phased.haps,phased.sample,log}") into phased
script:
def (bed, bim, fam) = input_bed
def (haplotype, legend, sample) = input_ref
"""
shapeit2 --thread "${task.cpus}" --input-bed "${bed}" "${bim}" "${fam}" --input-map "${input_map}" --input-ref "${haplotype}" "${legend}" "${sample}" --duohmm --output-max "${chrom}.phased" --seed 54321
"""
}
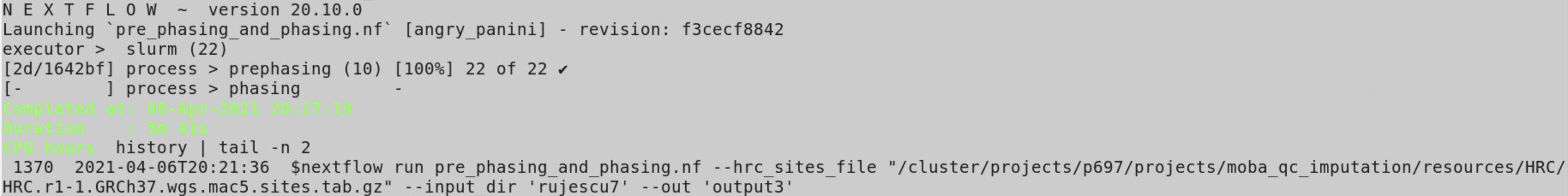
But getting no output so far, (attached image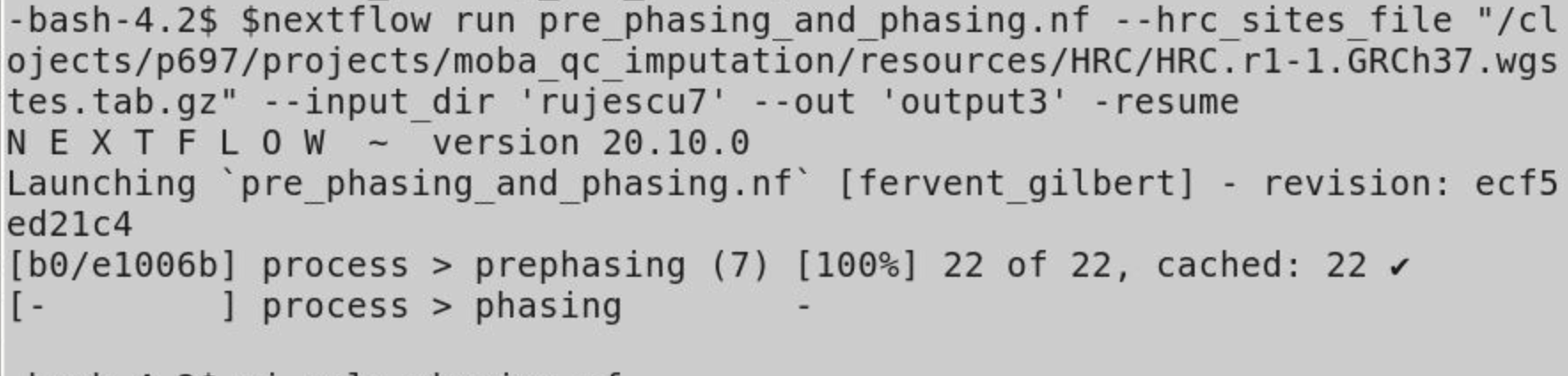 )
)
I have tried to check individual channel separately using channel.view(), identified the problems, modified the code and checked again. Using the code below I can see individual channel emits 22 lines [chr1-22,[file_list]]. After merging I can see [chr1-22,[joined_file_list].
channel
.fromFilePairs( "output3/*.{bed,bim,fam}", size:3 )
.set { plink_data }
plink_data.filter{chrom, bfiles -> bfiles.collect {it.getBaseName()}.every {it.endsWith("step10")}}.map{group_key,file_list -> tuple (group_key.
replaceFirst(/^*\.step10/,""), file_list)}.set {in_pl}
channel.fromFilePairs('/cluster/projects/p697/projects/moba_qc_imputation/resources/1000Genomes/genetic_map_chr*_combined_b37.txt',size:1).map {
group_key, file_list -> tuple(group_key.replaceFirst(/^*genetic_map_/,""),file_list.first())}.set {genetic_map}
//genetic_map.view()
channel.fromFilePairs('/cluster/projects/p697/projects/moba_qc_imputation/resources/HRC/haplegendsample/*hrc.r1-1.ega.grch37.chr*.haplotypes.{ha
p.gz,legend.gz,samples}',size:3).map {group_key,file_list -> tuple(group_key.replaceFirst(/^.*.hrc\.r1-1\.ega\.grch37\./,""),file_list)}.set {re
f_panel}
//ref_panel.view()
in_pl.join(ref_panel).join(genetic_map).into {shape_in;imp_in}
process phasing {
//publishDir params.out, mode:'copy'
input:
tuple val(chrom), path(input_bed), path(intput_ref), path(input_map) from shape_in
output:
tuple val("${chrom}"),path("${chrom}.{phased.haps,phased.sample,log}") in
to phased
script:
def (bed, bim, fam) = input_bed
def (haplotype, legend, sample) = input_ref
"""
//shapeit="/cluster/projects/p697/projects/moba_qc_imputation/software/shap
eit"
shapeit --thread "${task.cpus}" --input-bed "${bed}" "${bim}" "${fam}" --input-map "${input_map}" --input-ref "${haplotype}" "${legend}" "${samp
le}" --duohmm --output-max "${chrom}.phased" --seed 54321
"""
}
 But now I get error:
But now I get error:
No such variable: chrom
But I decalred "chrom" in the input
The input bed|bim|fam files from channel from FilePairs works but from a output channel of previous process does not. If I run phasing solo and all the inputs from channel from FilePairs it works. But if I run prephasing and phasing together, the prephasing process works but for phasing process not output and no error; Not sure what I am doing wrong.
hrc_sites_file = file(params.hrc_sites_file)
Channel.fromFilePairs( "${params.input_dir}/*.{bed,bim,fam}", size:3 ).set { plink_data }
Channel.of(1..22).set { chroms }
params.out='output/'
process prephasing {
publishDir params.out, mode:'copy'
input:
tuple val(prefix), path(bfiles) from plink_data
each chrom from chroms
path hrc_sites_file
output:
tuple val("${chrom}"),path("chr${chrom}*.{bed,bim,fam}") into qc1_chr
tuple val("${chrom}"),path("chr${chrom}*log") into qc1_log
tuple val("${chrom}"),path("*-chr${chrom}.step4-HRC.txt") into qc1_hrc
path("chr${chrom}.step1b.snplist.txt") into qc1_snps
path("chr${chrom}.step1a.dupvar") into qc1_dups
"""
PRE_PASHING_QC2.sh "${hrc_sites_file}" "${prefix}" "${chrom}"
"""
}
channel.fromFilePairs('/cluster/projects/p697/projects/moba_qc_imputation/resources/1000Genomes/genetic_map_chr*_combined_b37.txt',size:1).map {group_key, file_list -> tuple(group_key.replaceFirst(/^*gen
etic_map_/,""),file_list.first())}.set {genetic_map}
channel.fromFilePairs('/cluster/projects/p697/projects/moba_qc_imputation/resources/HRC/haplegendsample/*hrc.r1-1.ega.grch37.chr*.haplotypes.{hap.gz,legend.gz,samples}',size:3).map{group_key,file_list ->
tuple(group_key.replaceFirst(/^.*.hrc\.r1-1\.ega\.grch37\./,""),file_list)}.set {ref_panel}
qc1_chr.filter{chrom, bfiles -> bfiles.collect {it.getBaseName()}.every {it.endsWith("step10")}}.map{group_key,file_list -> tuple (group_key.replaceFirst(/^*\.step10/,""), file_list)}.join(ref_panel).joi
n(genetic_map).into {shape_in;imp_in}
imp_in.view()
process phasing {
publishDir params.out, mode:'copy'
input:
tuple val(chrom), path(input_bed), path(input_ref), path(input_map) from shape_in
output:
tuple val("${chrom}"),path("${chrom}.*") into phased
script:
def (bed, bim, fam) = input_bed
def (haplotype, legend, sample) = input_ref
"""
shapeit --thread "${task.cpus}" --input-bed "${bed}" "${bim}" "${fam}" --input-map "${input_map}" --input-ref "${haplotype}" "${legend}" "${sample}" --duohmm --output-max "${chrom}.step10" --seed 54321
"""
}