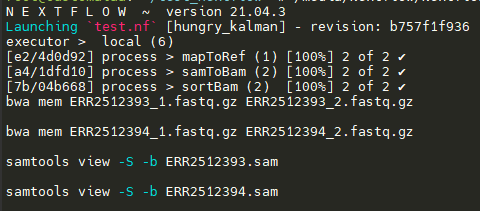
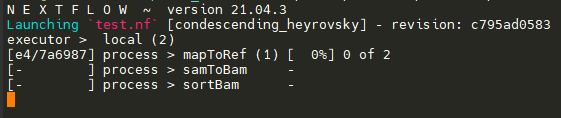
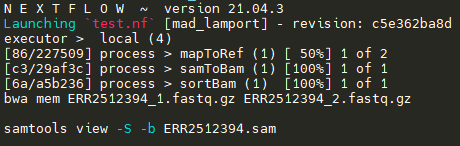
I have written a Nextflow script with three process:
- The first process takes a pair of
fastqfiles and aligns with reference genome. The process writes the resultingSAMfile intosam channel. - Second process takes input from the
sam channeland creates aBAMfile from it, and writes it intobam channel. - Final process reads from
bam channeland sorts theBAMfiles.
Following is the entire code:
#!~/bin nextflow
reads_ch = Channel.fromFilePairs('raw/*_{1,2}.fastq.gz', flat: true)
ref_genome = file('reference/genome/index/ref_genome')
process mapToRef {
memory '20 GB'
cpus 16
input:
tuple val(sample_id), file(read1), file(read2) from reads_ch
output:
tuple val(sample_id), file("${sample_id}.sam") into sam_ch
script:
"""
~/biotools/bwa/bwa mem -t 60 ${ref_genome} ${read1} ${read2} > ${sample_id}.sam
"""
}
process samToBam {
memory '20 GB'
cpus 16
input:
tuple val(sample_id), file("${sample_id}.sam") from sam_ch
output:
tuple val(sample_id), file("${sample_id}.bam") into bam_ch
script:
"""
~/biotools/SAMTOOLS/samtools-1.12/samtools view -S -b ${sample_id}.sam > ${sample_id}.bam
"""
}
process sortBam {
memory '20 GB'
cpus 16
input:
tuple val(sample_id), file("${sample_id}.bam") from bam_ch
output:
tuple val(sample_id), file("${sample_id}.sorted.bam") into sorted_bam_ch
script:
"""
~/biotools/SAMTOOLS/samtools-1.12/samtools sort ${sample_id}.bam -o ${sample_id}.sorted.bam
"""
}
All three processes successfully execute their commands. However, they are executed sequentially. For example, the first process mapToRef runs sequentially for all read pairs, followed by samToBam, which too, runs sequentially after all mapToRefs have finished execution. I was under impression that parallelization would "work" out of the box, but I must be doing something wrong. I am a complete beginner in Nextflow and would greatly appreciate any help regarding this.
EDIT
Output of reads_ch.view():
[ERR2512393, /home/work/raw/ERR2512393_1.fastq.gz, /home/work/raw/ERR2512393_2.fastq.gz]
[ERR2512394, /home/work/raw/ERR2512394_1.fastq.gz, /home/work/raw/ERR2512394_2.fastq.gz]
[ERR2512391, /home/work/raw/ERR2512391_1.fastq.gz, /home/work/raw/ERR2512391_2.fastq.gz]
[ERR2512392, /home/work/raw/ERR2512392_1.fastq.gz, /home/work/raw/ERR2512392_2.fastq.gz]
[ERR2512390, /home/work/raw/ERR2512390_1.fastq.gz, /home/work/raw/ERR2512390_2.fastq.gz]