You can use GSA.read.gmt function from GSA package. The following code can be used to convert the file to a dataframe. Just ignore the warnings.
Original_response
library(GSA)
data <- GSA.read.gmt("c5.all.v6.2.symbols.gmt")
gene_names <- unlist(data$genesets, use.names=FALSE)
your_dataframe <- cbind(data$geneset.names,gene_names)
colnames(your_dataframe) <- c("Pathways","Genes")
mydataframe <- as.data.frame(your_dataframe)
head(mydataframe)
Pathway Gene
1 GO_POSITIVE_REGULATION_OF_VIRAL_TRANSCRIPTION POLR2C
2 GO_CARDIAC_CHAMBER_DEVELOPMENT POLR2J
3 GO_DNA_DEPENDENT_DNA_REPLICATION_MAINTENANCE_OF_FIDELITY CTDP1
4 GO_CIRCADIAN_RHYTHM RDBP
5 GO_PHOSPHATIDYLSERINE_ACYL_CHAIN_REMODELING COBRA1
6 GO_SPINAL_CORD_DEVELOPMENT RSF1
Edited_response
You can use the code given below to achieve your desired output:
library(GSA)
data <- GSA.read.gmt("c5.all.v6.2.symbols.gmt")
len_vec=c() # Now create a vector for containing the length of genes at each position
len_vec[1] = 3
for(i in 1:length(data$genesets)){len_vec[i] <- c(length(data$genesets[[i]]))}
pathway_vec <- unlist(Vectorize(rep.int)(data$geneset.names, len_vec),use.names = FALSE) # Now create a vector for all the pathways in the data
desired_df <- as.data.frame(cbind(pathway_vec,unlist(data$genesets,use.names = FALSE))) # This gives your desired dataframe
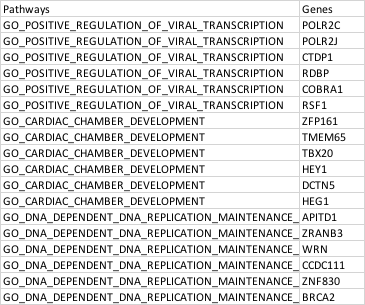
head(desired_df)
Pathway Gene
1 GO_POSITIVE_REGULATION_OF_VIRAL_TRANSCRIPTION POLR2C
2 GO_POSITIVE_REGULATION_OF_VIRAL_TRANSCRIPTION POLR2J
3 GO_POSITIVE_REGULATION_OF_VIRAL_TRANSCRIPTION CTDP1
4 GO_POSITIVE_REGULATION_OF_VIRAL_TRANSCRIPTION RDBP
5 GO_POSITIVE_REGULATION_OF_VIRAL_TRANSCRIPTION COBRA1
6 GO_POSITIVE_REGULATION_OF_VIRAL_TRANSCRIPTION RSF1