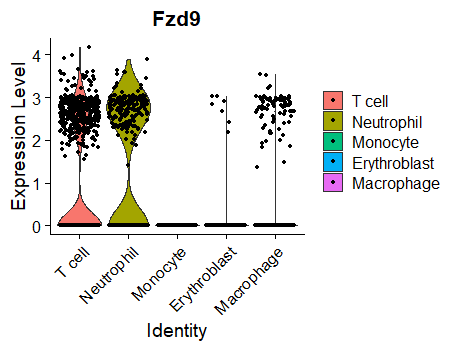
Seurat VlnPlots are most commonly used to visualize differences in any given gene expression across multiple clusters or cell types. For example:
VlnPlot(object = MouseCellAtlas, idents = c("T cell","Neutrophil","Erythroblast","Monocyte","Macrophage"), features = 'Fzd9')
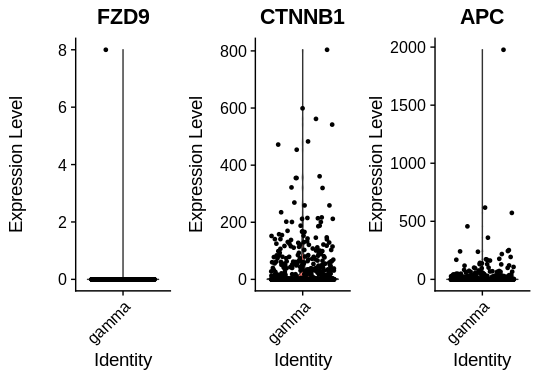
But suppose I want to view the expression of several genes in just idents = "T cell". I can do this to generate three separate plots:
VlnPlot(object = mca, idents = 'T cell', features = c('Fzd9','Ctnnb1','Apc'), combine = TRUE, ncol = 3)
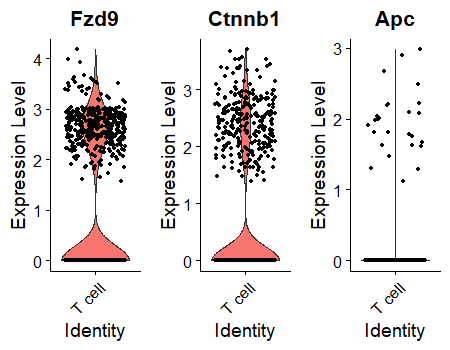
But ideally I'd like this second plot to look just like the first plot, except with only a single tissue on the legend ("T cell") and multiple genes in the various colored violins. Unfortunately, I am not aware of any simple way to combine these violins into a single plot area.
This doesn't do what I need:
VlnPlot(object = mca, idents = 'T cell', features = c('Fzd9','Ctnnb1','Apc'), combine = TRUE, ncol = 3)
Is there built-in functionality in Seurat for generating a VlnPlot for multiple genes in a single tissue in a single plot? Or can anyone suggest a workaround?