I have surprisingly low counts when running featureCounts on some (single-end) RNA-seq data mapped on C. elegans genome using hisat2.
To more easily show the problem, I generated a small subset of the bam file and of the annotation file I'm using. Here is what I can see when loading these two files on IGV:
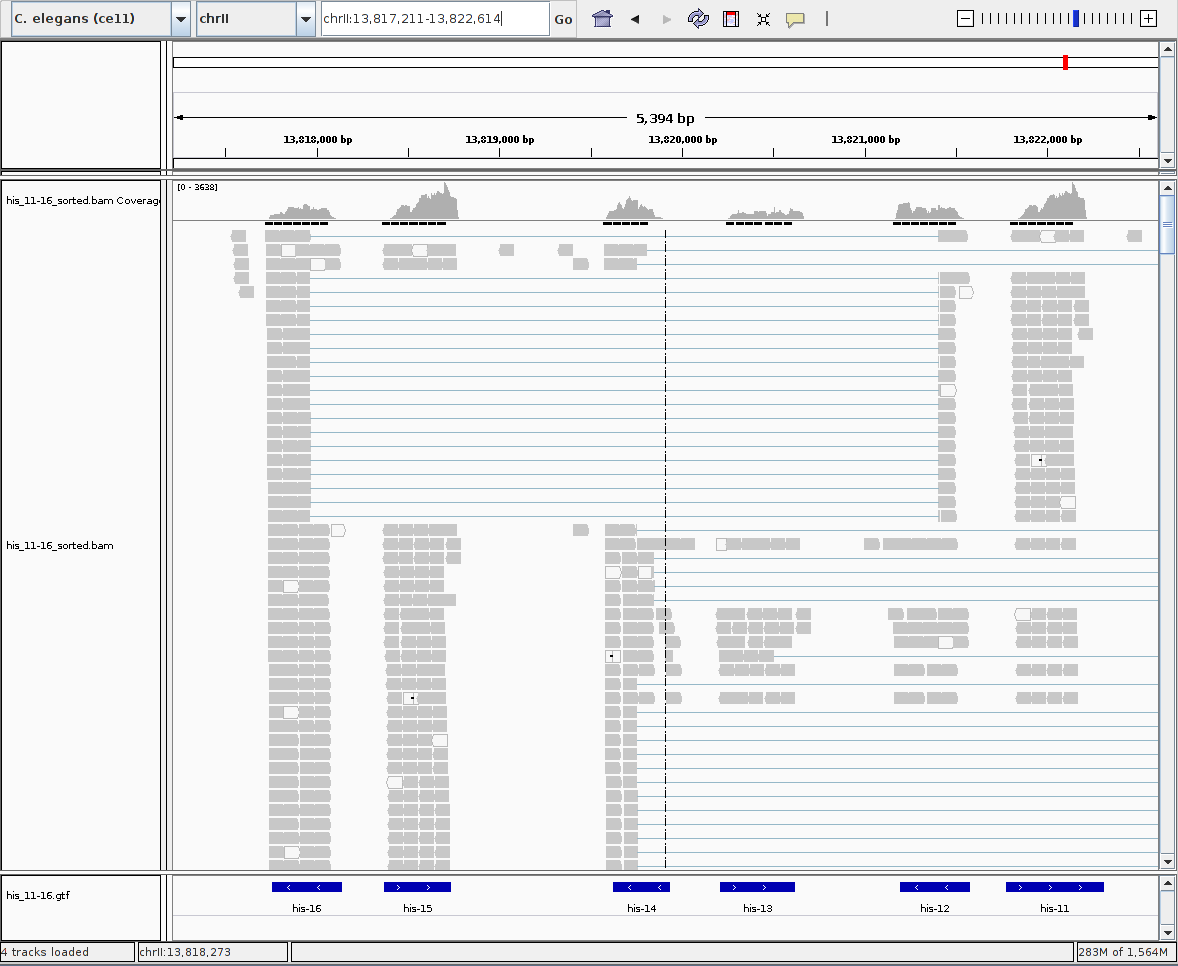
The coverage for the "his-11" and "his-15" genes peak at around 3600. For "his-12" and "his-16", it peaks at more than 1000
Here are the content of my annotation file:
$ cat his_11-16.gtf
II ensembl transcript 13817759 13818143 . - . gene_biotype "protein_coding"; gene_id "WBGene00001890"; gene_name "his-16"; gene_source "ensembl"; gene_version "1"; p_id "P8354"; transcript_biotype "protein_coding"; transcript_id "ZK131.10"; transcript_name "ZK131.10"; transcript_source "ensembl"; transcript_version "1"; tss_id "TSS11539";
II ensembl transcript 13818371 13818739 . + . gene_biotype "protein_coding"; gene_id "WBGene00001889"; gene_name "his-15"; gene_source "ensembl"; gene_version "1"; p_id "P8185"; transcript_biotype "protein_coding"; transcript_id "ZK131.9"; transcript_name "ZK131.9"; transcript_source "ensembl"; transcript_version "1"; tss_id "TSS52436";
II ensembl transcript 13819626 13819937 . - . gene_biotype "protein_coding"; gene_id "WBGene00001888"; gene_name "his-14"; gene_source "ensembl"; gene_version "1"; p_id "P21890"; transcript_biotype "protein_coding"; transcript_id "ZK131.8"; transcript_name "ZK131.8"; transcript_source "ensembl"; transcript_version "1"; tss_id "TSS50308";
II ensembl transcript 13820210 13820620 . + . gene_biotype "protein_coding"; gene_id "WBGene00001887"; gene_name "his-13"; gene_source "ensembl"; gene_version "1"; p_id "P2149"; transcript_biotype "protein_coding"; transcript_id "ZK131.7"; transcript_name "ZK131.7"; transcript_source "ensembl"; transcript_version "1"; tss_id "TSS35106";
II ensembl transcript 13821198 13821581 . - . gene_biotype "protein_coding"; gene_id "WBGene00001886"; gene_name "his-12"; gene_source "ensembl"; gene_version "1"; p_id "P26082"; transcript_biotype "protein_coding"; transcript_id "ZK131.6"; transcript_name "ZK131.6"; transcript_source "ensembl"; transcript_version "1"; tss_id "TSS23693";
II ensembl transcript 13821778 13822314 . + . gene_biotype "protein_coding"; gene_id "WBGene00001885"; gene_name "his-11"; gene_source "ensembl"; gene_version "1"; p_id "P10535"; transcript_biotype "protein_coding"; transcript_id "ZK131.5"; transcript_name "ZK131.5"; transcript_source "ensembl"; transcript_version "1"; tss_id "TSS52792";
The featureCounts run:
$ featureCounts -a his_11-16.gtf -o his_11-16_counts.txt -t transcript -g gene_name -O his_11-16_sorted.bam
========== _____ _ _ ____ _____ ______ _____
===== / ____| | | | _ \| __ \| ____| /\ | __ \
===== | (___ | | | | |_) | |__) | |__ / \ | | | |
==== \___ \| | | | _ <| _ /| __| / /\ \ | | | |
==== ____) | |__| | |_) | | \ \| |____ / ____ \| |__| |
========== |_____/ \____/|____/|_| \_\______/_/ \_\_____/
v1.5.2
//========================== featureCounts setting ===========================\\
|| ||
|| Input files : 1 BAM file ||
|| S his_11-16_sorted.bam ||
|| ||
|| Output file : his_11-16_counts.txt ||
|| Summary : his_11-16_counts.txt.summary ||
|| Annotation : his_11-16.gtf (GTF) ||
|| Dir for temp files : ./ ||
|| ||
|| Threads : 1 ||
|| Level : meta-feature level ||
|| Paired-end : no ||
|| Strand specific : no ||
|| Multimapping reads : not counted ||
|| Multi-overlapping reads : counted ||
|| Min overlapping bases : 1 ||
|| ||
\\===================== http://subread.sourceforge.net/ ======================//
//================================= Running ==================================\\
|| ||
|| Load annotation file his_11-16.gtf ... ||
|| Features : 6 ||
|| Meta-features : 6 ||
|| Chromosomes/contigs : 1 ||
|| ||
|| Process BAM file his_11-16_sorted.bam... ||
|| Single-end reads are included. ||
|| Assign reads to features... ||
|| Total reads : 35037 ||
|| Successfully assigned reads : 1849 (5.3%) ||
|| Running time : 0.00 minutes ||
|| ||
|| Read assignment finished. ||
|| ||
|| Summary of counting results can be found in file "his_11-16_counts.txt" ||
|| ||
\\===================== http://subread.sourceforge.net/ ======================//
And the resulting counts file:
$ cat his_11-16_counts.txt
# Program:featureCounts v1.5.2; Command:"featureCounts" "-a" "his_11-16.gtf" "-o" "his_11-16_counts.txt" "-t" "transcript" "-g" "gene_name" "-O" "his_11-16_sorted.bam"
Geneid Chr Start End Strand Length his_11-16_sorted.bam
his-16 II 13817759 13818143 - 385 869
his-15 II 13818371 13818739 + 369 3
his-14 II 13819626 13819937 - 312 953
his-13 II 13820210 13820620 + 411 23
his-12 II 13821198 13821581 - 384 423
his-11 II 13821778 13822314 + 537 1
Why such a low assignment rate, why so little counts?
I see that hisat2 decided to split an important proportion of the reads between orthologs, like "his-16" and "his-12", but my understanding is that the default values for the minOverlap and fracOverlap parameters should ensure that such split reads will be counted:
--minOverlap <int> Minimum number of overlapping bases in a read that is
required for read assignment. 1 by default. Number of
overlapping bases is counted from both reads if paired
end. If a negative value is provided, then a gap of up
to specified size will be allowed between read and the
feature that the read is assigned to.
--fracOverlap <float> Minimum fraction of overlapping bases in a read that is
required for read assignment. Value should be within range
[0,1]. 0 by default. Number of overlapping bases is
counted from both reads if paired end. Both this option
and '--minOverlap' option need to be satisfied for read
assignment.
What did I get wrong?