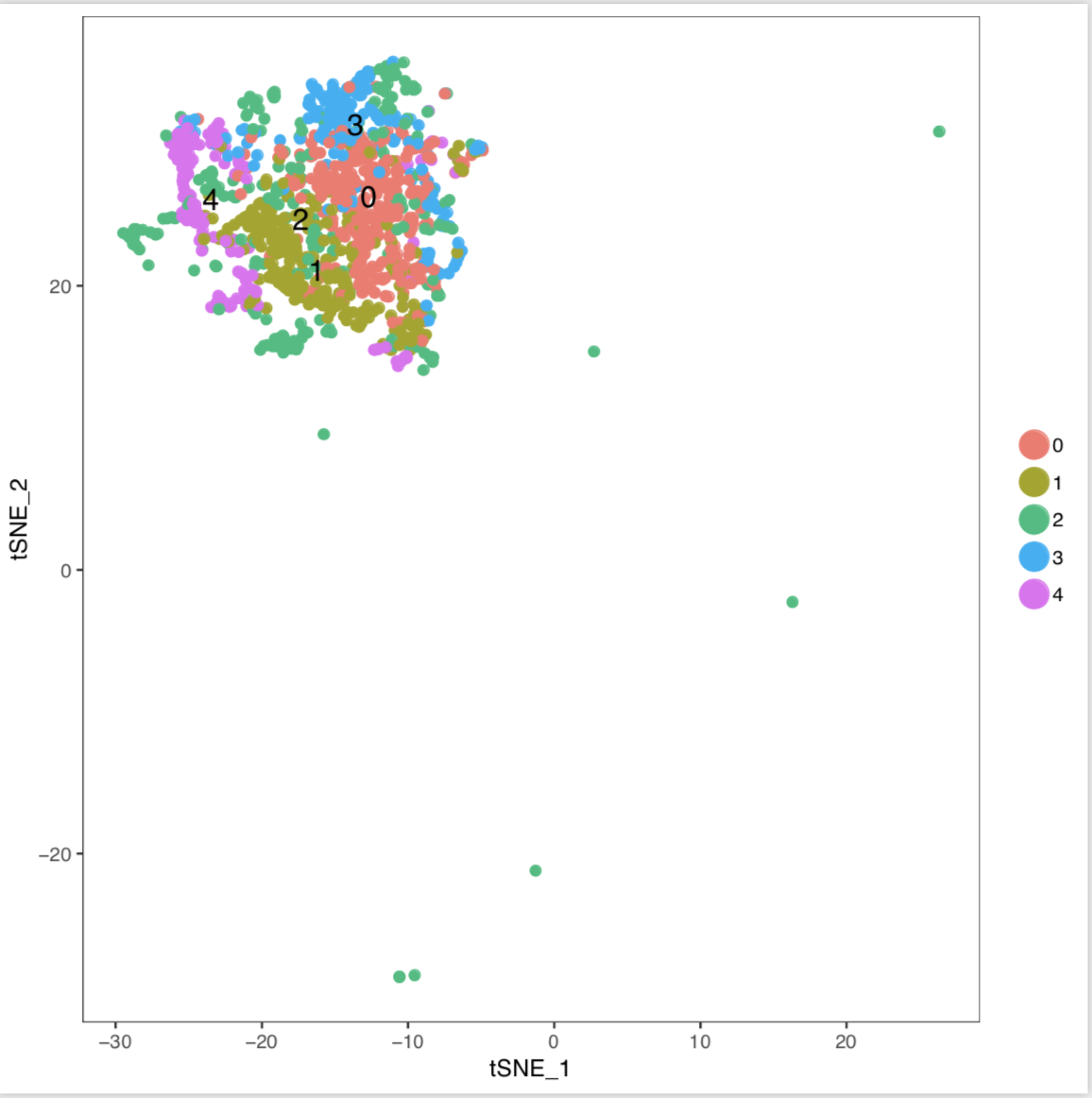
I have no idea why I used the subsetdata in Seurat after using CCA the TSNEPlot shows the cluster all in the left side and merge, did I miss something? Here are the code and figure I got.
cluster <-SubsetData(object = immune.combined, ident.use= "3")
cluster <- NormalizeData(object = cluster, normalization.method = "logNormalize", scale.factor = 10000)
cluster <- FindVariableGenes(object = cluster, mean.function = ExpMean, dispersion.function = LogVMR, x.low.cutoff = 0, x.high.cutoff = 6, y.cutoff = 0, do.plot = F)
cluster <- ScaleData(object = cluster, display.progress = FALSE)
cluster <- RunPCA(object = cluster, pc.genes = [email protected], pcs.print = 1:10, genes.print =5)
PCAPlot(object = cluster, group.by= "orig.ident", dim.1 = 1, dim.2 = 2)
cluster <- FindClusters (object = cluster, reduction.type = "pca", dims.use = 1:10, resolution =0.3, print.output = 0, save.SNN = TRUE, force.recalc = T)
TSNEPlot(object = cluster, do.label = TRUE,do.return = TRUE,pt.size = 2,label.size = 5, no.legend=FALSE)