I have some highly contaminated ancient DNA sequences. I have adapter removed and collapsed these and run them through Kraken2. The Kraken reports show multiple levels of taxa with good numbers of reads in each.
However, when I run Bracken - I keep getting an error saying it can't find any reads.
Error: no reads found. Please check your Kraken report
I have tried adjusting the taxonomic level (all the way up to Phylum) and get the same result. I have tried using different databases to run the Kraken analysis through and updating both packages.
Any ideas what could be going wrong?
Update:
Here's the head of one of the report files:
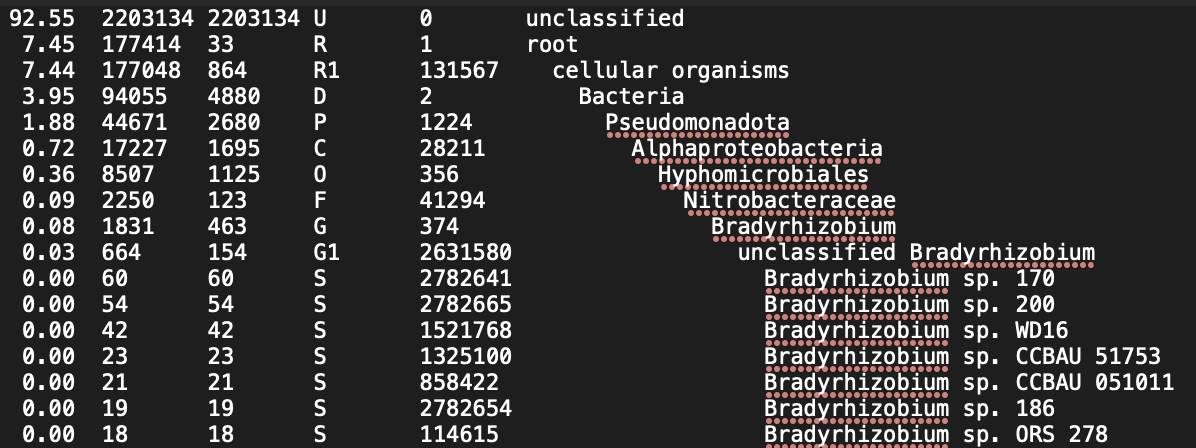
I am also using the latest version of Bracken downloaded via Bioconda. My command to run Bracken is via a bash script:
#SBATCH --partition=day
#SBATCH --output=slurm_bracken_job%J.out
#SBATCH --mem-per-cpu=10G
#SBATCH --cpus-per-task=32
# we load kraken2 and bracken into our environment:
ml kraken2
ml bracken
NAME=$1
srun bracken -d /workspaces/groups/database/nt-taxonomy-2021-02-04_braken/ -i ${NAME}.kreport -o ${NAME}.bracken -r 75 -l S
This is then run using:
for i in $(ls -1 ./*.report | sed 's/.report//'); do sbatch Test_Kraken.sh "$i"; done
bracken -d /workspaces/groups/database/nt-taxonomy-2021-02-04_braken/ -i Sample_1.collapsed.report -o Sample_!.report.bracken -r 75 -l Sand still the same error $\endgroup$