We cannot assume that doublets will produce more UMIs
I would caution the assumption that all doublets will have twice the UMI levels of isolated single-cells. Many "doublets" could contain multiplets of 3 or more cells, depending on how many cells have been loaded into the experiment. Most vendors of single-cell technologies quote multiplet rates for doublets, triplets, etc combined.
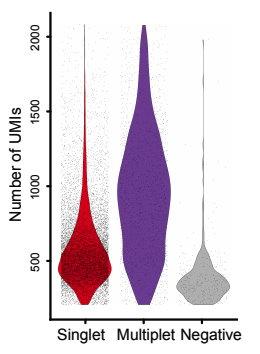
Furthermore, we cannot assume that the data from multiplet (doublet) cells is of high quality or produces comparable reads and UMI counts than we would expect from two single cells combined. Some tests of doublets (such as using mixed mouse and human samples) have found that the data generated from doublet droplets or nanowells has relatively low UMI abundance. This may be due to the protocols having been optimised for droplets or nanowells containing only one cell. Especially where tissue preparation is required this is a concern and is consistent with observations that doublet cells appear to be broken or contaminated with debris when they can be identified. This is highly dependent on the tissue you and working with, sample preparation, and the platform used to separate single-cells.
I have not been able to reproduce the above result using data from various species (using total UMI counts per droplet or housekeeper genes), although this is work still in progress. Even in the data shown, many multiplet samples have similar total UMI counts to the true single-cells. Therefore, they cannot be separated by a simple threshold (despite being significantly different). There have been several tools developed already for detecting multiplets (and filtering them out) but this is still an area of active development and there are more ways that these could be refined. In many single-cell analyses, arbitrary thresholds are being used to filter the data before further analysis and this may be inadvertently reducing the impact of multiplets if they have produced fewer UMI counts than single-cells for which the protocols have been optimised for.
Experimental protocols can be optimised to reduce doublets
If you have any insight into how these bioinformatics solutions to issues with multiplets could be used or improved I encourage you to pursue it. However, I suspect that the eventual solution to this issue is in optimising the single-cell NGS experiments for the sample type and platform that you are working with. Technology in single-cell 'omics is continuing to develop and any solution which produces more useful single-cell data than contaminants or doublets (that need to be removed), the more cells can be sampled at the same cost. There is a strong incentive for experimentalists and single-cell technology vendors to optimise this issue.