I am running some analysis on an RNA-seq dataset. I have a list of transcripts that are potential lncRNA for which I ran both Kallisto and Salmon aligners. The input data for index building and quantification includes mRNA as well as these potential lncRNA.
Commands for one replicate:
Salmon
salmon index --keepDuplicates -t transcripts.fasta -i index_directory/ -k 31
salmon quant --seqBias --gcBias -i index_directory/ --libType ISR --auxDir index_directory/ -1 sample_R1.fastq.gz -2 sample_R2.fastq.gz -o output_name
Kallisto
kallisto index -i kallisto_index.idx transcripts.fasta
kallisto quant -i kallisto_index.idx -o output_name --rf-stranded sample_R1.fastq.gz sample_R2.fastq.gz
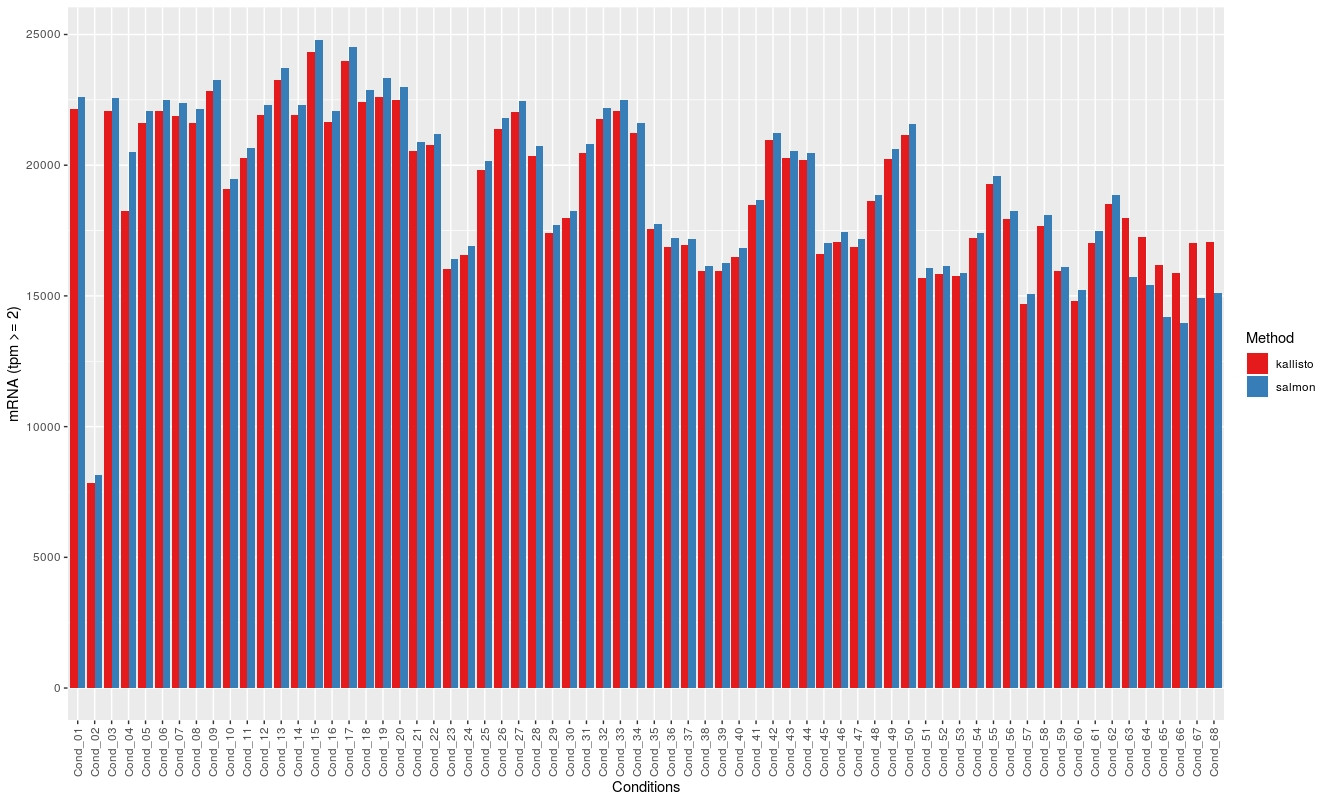
Then I used the tpm values, averaged replicates (3) and plotted the number of transcripts per condition that had a tpm > 2. Same result for tpm > 5.
The following graph shows the for mRNA:
And for lncRNA:
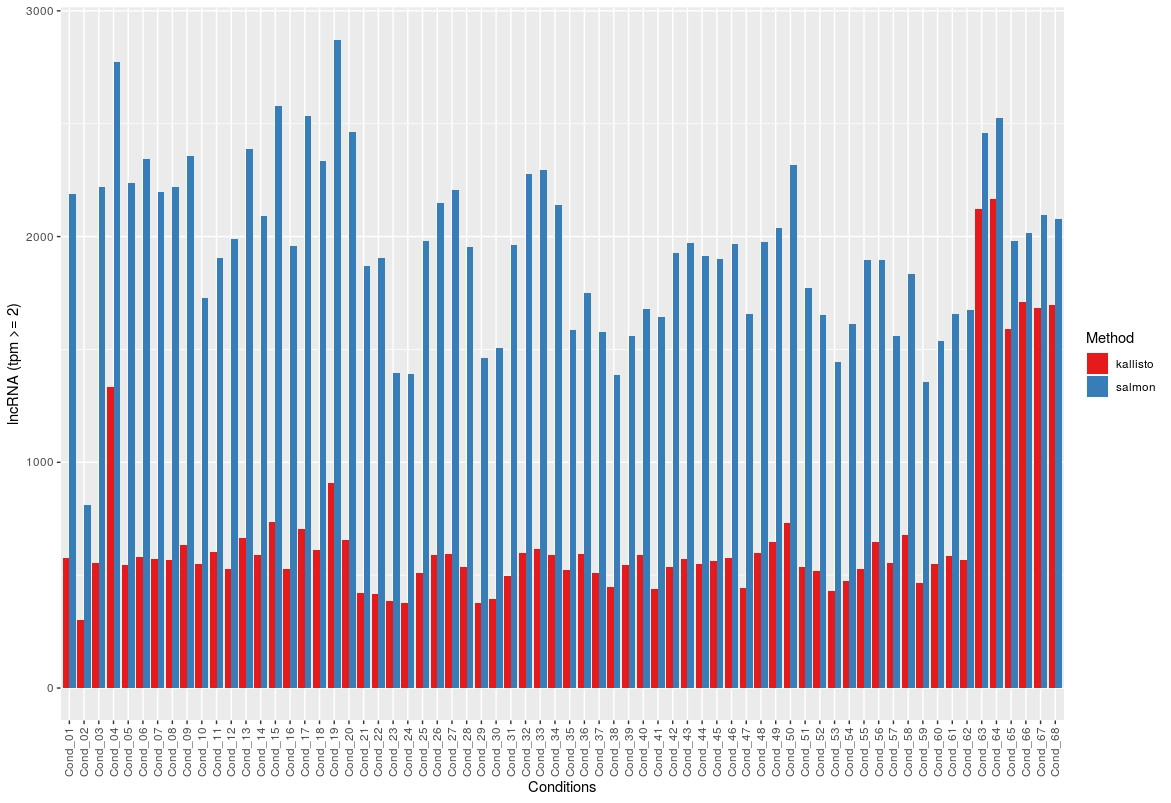
Again, the analysis were done with both lncRNA and mRNA transcripts together in the same fasta file.
The overlap is not shown here but is always smaller than the count for Kallisto, for the lncRNA.
In literature both tools seem to have similar results. I cannot find a reason for this. If there would be an error somewhere I would expect to see also differences in the conding RNAs which does not happen.