I have created a ComplexHeatmap containing 2 Heatmaps, each with their own legend. The legend on the first Heatmap is continuous, whereas that of the second is discrete.
When I concatenate these Heatmaps vertically, and draw the HeatmapList, their legends are auto-aligned to the center of the plot. Is there any way I can have these legends positioned centered to the respective heatmaps and not to the center of the plot?
This is what I have:
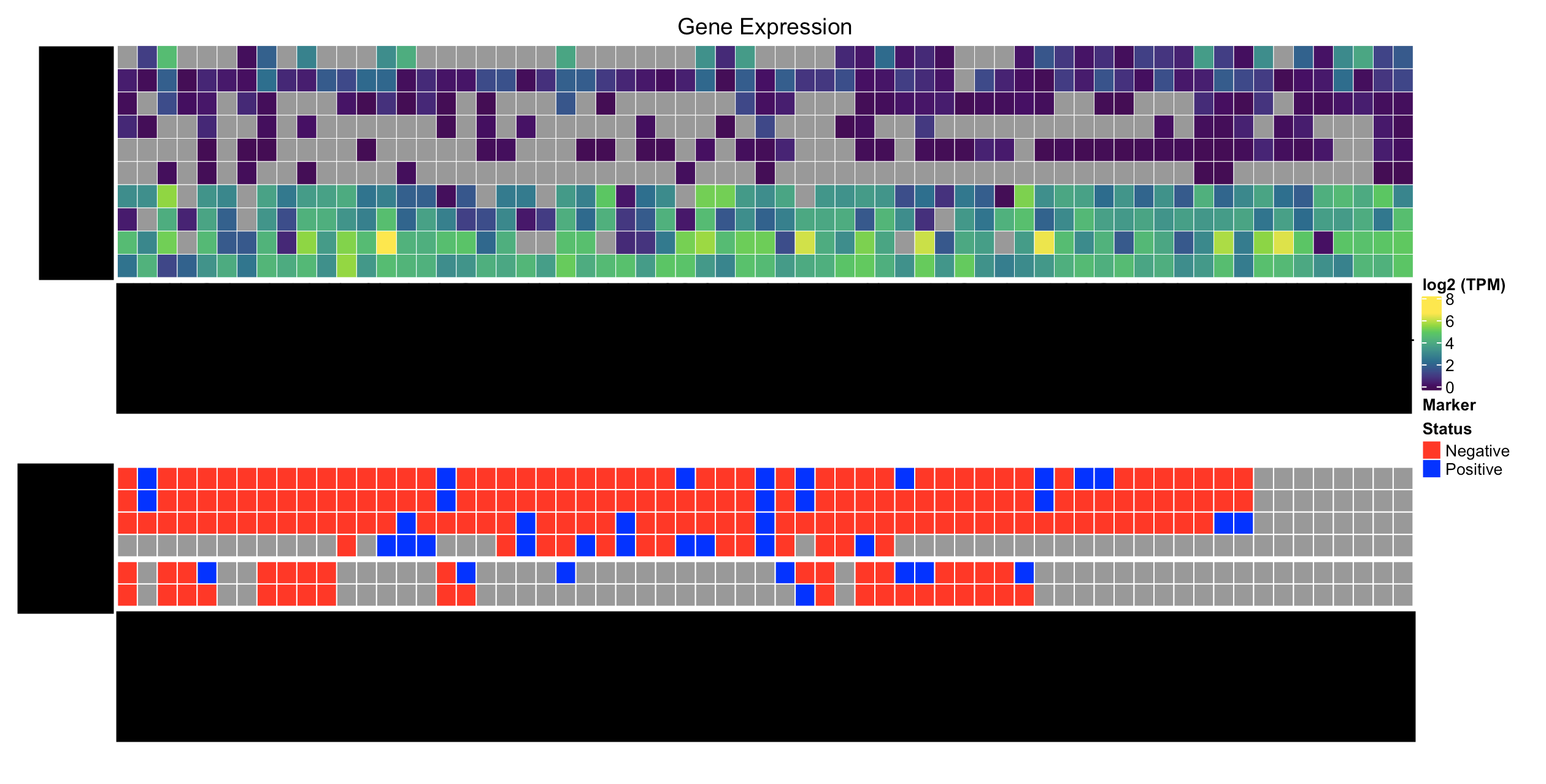
Here's what I'd like to have:
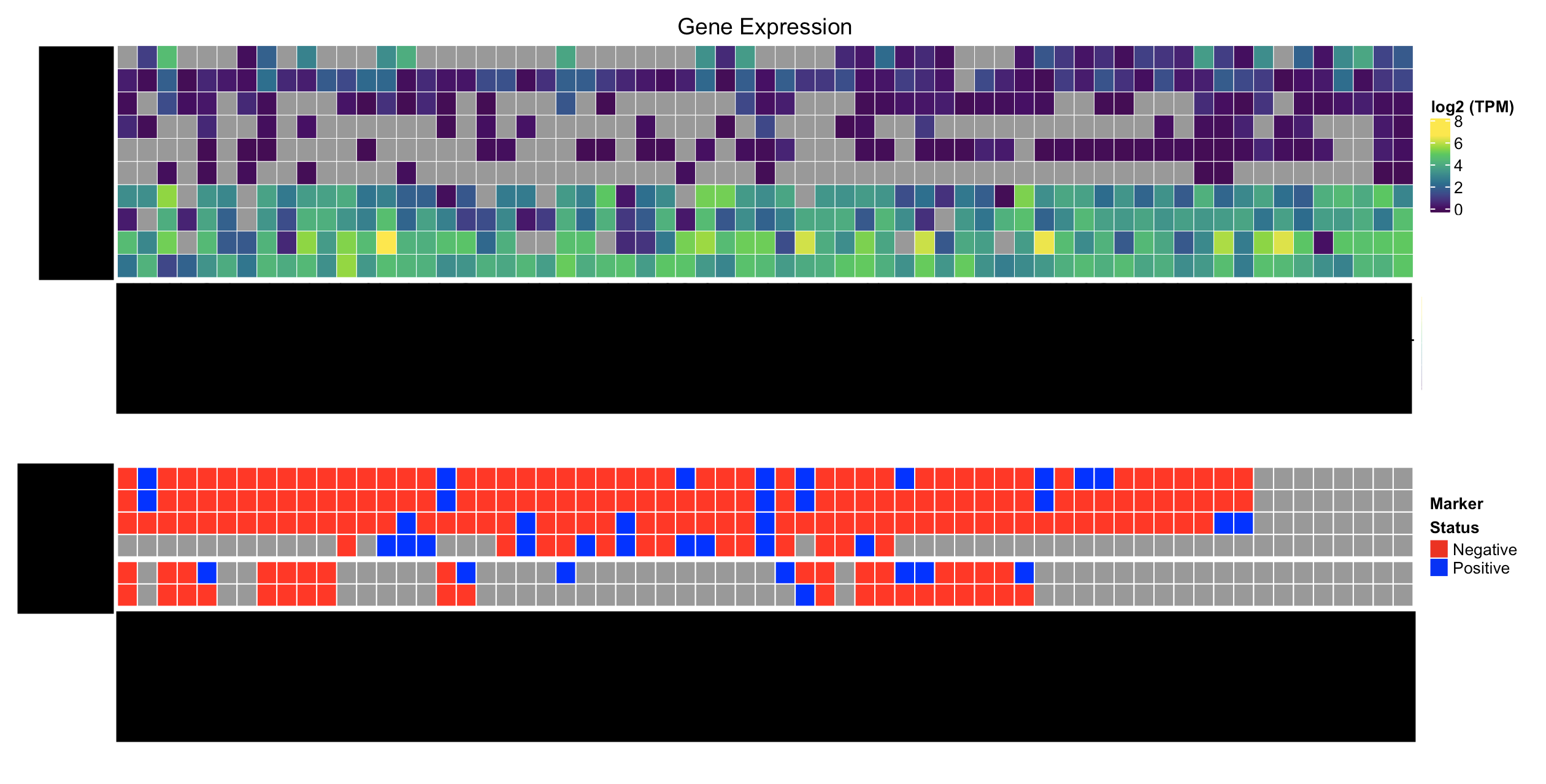
How can I go about solving this problem? I'd really like to avoid creating customized Legends if I can, but if that's what it takes, that works too.
Thank you in advance for any help/pointers!
Note: I tried adding the ComplexHeatmap tag, but it doesn't exist yet and I don't have the reputation to create new tags yet.
Edit: Since this is a Bioconductor package, I also created an identical post on the Bioconductor support forum
Dummy data and code:
devtools::install_github('jokergoo/ComplexHeatmap');
library(ComplexHeatmap);
library(viridis);
dummy_top_mat <- structure(c(0.4563, 0.2211, 1.2235, 0.067, 1.6859, 1.0936, 0.4533,
1.6844, 1.0039, 0.5402, 0.6841, 1.498, 1.042, 0.1711, 0.3565,
2.2814, 3.0516, 1.5012, 0.5367, 3.0846, 1.1909, 0.235, 1.4266,
0.4858, 2.9054, 0.3733, 0.6902, 0.8555, 0.4234, 2.6778, 0.1568,
0.5556, 2.0172, 0.8034, 2.2897, 0.1166, 3.8033, 0.1431, 2.0606,
1.2725, 1.5365, 0.4123, 1.2087, 1.1264, 0.8334, 1.1943, 1.58,
1.5849, 0.3004, 0.3722, 0.0362, 0.0532, 1.4867, 0.4053, 0.3615,
0.0897, 1.3217, 1.1447, 1.3058, 0.1903, 0.1067, 0.9482, 1.3382,
3.2955, 0.391, 1.0418, 0.2041, 1.208, 1.5857, 3.5313, 0.472,
1.389, 0.2143, 0.0226, 0.029, 0.444, 2.0521, 0.3955, 0.3495,
0.5062, 1.3236, 1.3234, 0.7111, 0.1176, 2.2223, 1.2073, 0.3964,
2.1175, 0.3382, 0.2816, 0.71, 3.1417, 0.2402, 0.5793, 0.7662,
1.6782, 0.0986, 0.087, 0.5447, 2.6672, 1.2498, 1.0676, 1.8608,
1.8146, 0.1422, 0.4221, 0.0303, 0.9541, 0.7358, 1.7664, 1.5144,
0.2034, 0.9366, 0.7837, 0.3284, 0.1477, 1.8306, 1.3564, 0.1126,
0.0171, 2.9858, 0.0233, 0.2796, 0.6995, 1.6081, 0.215, 1.7093,
0.5178, 1.7061, 2.473, 1.8912, 0.7661, 4.4102, 1.2963, 0.6542,
0.4281, 0.4491, 0.6, 0.4076, 1.6869, 0.4747, 3.9823, 1.1226,
2.7355, 2.7036, 0.2241, 0.983, 1.0992, 1.4736, 0.1584, 0.2995,
0.2272, 0.5744, 0.9314, 0.6924, 0.0812, 1.361, 0.727, 0.1525,
1.3367, 0.566, 2.7801, 0.2349, 3.2655, 1.0675, 0.449, 0.5411,
0.8291, 0.52, 0.0507, 0.6538, 0.4636, 1.2063, 0.9784, 0.1925,
0.0756, 0.136, 1.2529, 0.317, 0.0281, 0.8668, 3.4138, 5.3898,
0.521, 0.087, 3.4819, 0.1114, 0.0061, 0.9804, 1.139, 1.4159,
1.0297, 0.6503, 1.0828, 2.3527, 0.057, 0.5602, 0.4017, 0.9985,
0.8673), .Dim = c(10L, 20L), .Dimnames = list(c("gene01", "gene02",
"gene03", "gene04", "gene05", "gene06", "gene07", "gene08", "gene09",
"gene10"), c("sample01", "sample02", "sample03", "sample04",
"sample05", "sample06", "sample07", "sample08", "sample09", "sample10",
"sample11", "sample12", "sample13", "sample14", "sample15", "sample16",
"sample17", "sample18", "sample19", "sample20")));
dummy_bottom_mat <- structure(c("Positive", "Positive", "Positive", "Positive", "Negative",
"Positive", "Positive", "Positive", "Positive", "Negative", "Positive",
"Positive", "Positive", "Positive", "Positive", "Negative", "Positive",
"Negative", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Negative", "Positive", "Positive", "Positive", "Positive", "Positive",
"Negative", "Positive", "Positive", "Positive", "Negative", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Negative",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive", "Negative", "Positive", "Positive", "Positive", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive", "Positive", "Positive", "Positive", "Positive", "Positive",
"Positive"), .Dim = c(6L, 20L), .Dimnames = list(c("marker1",
"marker2", "marker3", "marker4", "marker5", "marker6"), c("sample01",
"sample02", "sample03", "sample04", "sample05", "sample06", "sample07",
"sample08", "sample09", "sample10", "sample11", "sample12", "sample13",
"sample14", "sample15", "sample16", "sample17", "sample18", "sample19",
"sample20")));
hmap1 <- Heatmap(dummy_top_mat, col = colorRampPalette(viridis(15))(100), name = 'log2 (TPM)', na_col = 'grey60', cluster_rows = FALSE, cluster_columns = FALSE, row_names_side = 'left', column_title_side = 'top', rect_gp = gpar(col='white', lwd=0.5), height=unit(10*0.75,'cm'), width = unit(20*0.75, 'cm'), column_title = 'Gene Expression');
hmap2 <- Heatmap(dummy_bottom_mat, col = c('Negative'='red', 'Positive'='blue'), na_col = 'grey60', row_split = factor( c('gp1','gp1','gp1','gp1','gp2','gp2'), c('gp1','gp2'), ordered = TRUE), name = 'Marker\nStatus', rect_gp = gpar(col='white',lwd=1), row_title = NULL, height = unit(6*0.75,'cm'), width = unit(20*0.75, 'cm'), row_names_side = 'left', column_title = 'Biomarkers');
draw(hmap1 %v% hmap2, ht_gap = unit(1, 'cm'), auto_adjust = FALSE);
sessionInfo()
R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin13.4.0 (64-bit)
Running under: macOS 10.14.4
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Users/ram/miniconda3/lib/R/lib/libRblas.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid stats graphics grDevices utils datasets methods base
other attached packages:
[1] viridis_0.5.1 viridisLite_0.3.0 ComplexHeatmap_2.1.0 forcats_0.4.0
[5] stringr_1.4.0 dplyr_0.8.0.1 purrr_0.2.5 readr_1.3.1
[9] tidyr_0.8.3 tibble_2.1.1 ggplot2_3.1.1 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.0 lubridate_1.7.4 lattice_0.20-38 circlize_0.4.6 prettyunits_1.0.2
[6] png_0.1-7 ps_1.3.0 rprojroot_1.3-2 assertthat_0.2.1 digest_0.6.18
[11] R6_2.3.0 cellranger_1.1.0 plyr_1.8.4 backports_1.1.4 reprex_0.2.1
[16] httr_1.4.0 pillar_1.3.1 GlobalOptions_0.1.0 rlang_0.3.1 lazyeval_0.2.2
[21] readxl_1.3.1 rstudioapi_0.10 callr_3.2.0 GetoptLong_0.1.7 desc_1.2.0
[26] devtools_2.0.2 munsell_0.5.0 broom_0.5.2 compiler_3.5.1 modelr_0.1.4
[31] pkgconfig_2.0.2 pkgbuild_1.0.3 shape_1.4.4 tidyselect_0.2.5 gridExtra_2.3
[36] crayon_1.3.4 withr_2.1.2 nlme_3.1-137 jsonlite_1.6 gtable_0.3.0
[41] magrittr_1.5 scales_1.0.0 cli_1.1.0 stringi_1.4.3 remotes_2.0.4
[46] fs_1.3.1 xml2_1.2.0 generics_0.0.2 rjson_0.2.20 RColorBrewer_1.1-2
[51] tools_3.5.1 glue_1.3.0 hms_0.4.2 processx_3.3.1 pkgload_1.0.2
[56] parallel_3.5.1 clue_0.3-57 colorspace_1.4-1 cluster_2.0.7-1 sessioninfo_1.1.1
[61] rvest_0.3.3 memoise_1.1.0 haven_2.1.0 usethis_1.5.0
draw(..., Legend(), ...)then we can use "just" argument withindraw()to define position. But if we want to adjust existing legend withinHeatmapfunction usingHeatmap(..., heatmap_legend_param = list(it doesn't have "just" argument. (See 5.4 Section of the manual). $\endgroup$just, it seems to be more along the lines of "where in the image" than "where with respect to the heatmap this legend belongs to". I will open a GitHub issue though, maybe the dev has a line of thought for this. $\endgroup$